Cell
cycle regulation and cancer
by R. Bernards
Cancer results from multiple genetic alterations in genes that control various aspects of
Many of the genes that are mutated in human cancer are directly involved in regulation of the cell division cycle, because such genes are most intimately linked to the machinery that controls cell proliferation.
A basic understanding of the machinery that drives the cell division cycle (or cell cycle, for short) is therefore indispensable for the study of molecular oncology.
Broadly, we can distinguish two groups of cancer genes: oncogenes and tumor suppressor genes.
In this lecture, I will discuss two major tumor suppressor pathways:
1. The p16-cyclin D-pRb-E2F pathway.
2. The p19ARF-Mdm2-p53 pathway.
Both pathways are frequently deregulated in human cancer and regulate the cell cycle machinery directly.
The oncogene and tumor suppressor pathways described here have in common that their gene products ultimately control transcription. This is perhaps not surprising since stable alterations in cellular behavior must be the consequence of alterations in gene expression patterns in the cell. For this reason, cancer-relevant genes often target gene expression directly to have such dramatic effects on cellular phenotype.
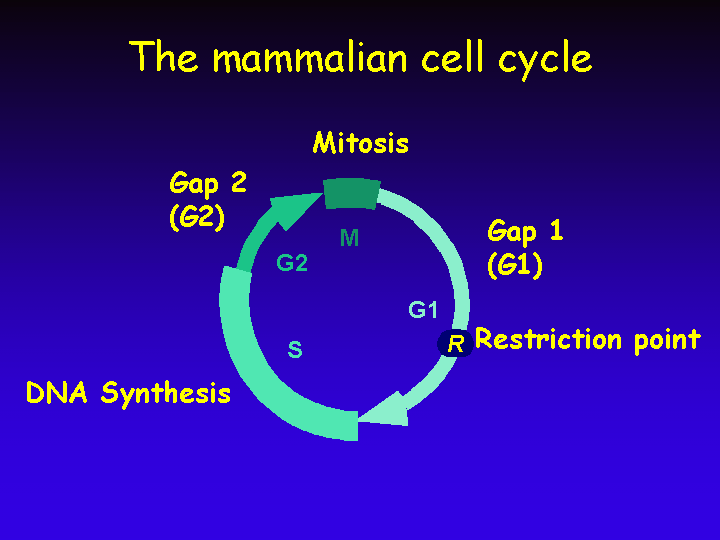
The Cell Cycle
The mammalian cell cycle consists of four discrete phases: S phase, in which DNA is replicated; M phase, in which the chromosomes are separated over two new nuclei in the process of mitosis. These two phases are separated by two so called “Gap” phases, G1 and G2, in which the cell prepares for the upcoming events of S and M, respectively.
A critical “checkpoint” in the mammalian cell cycle is the Restriction point (R).
Resting cells (the vast majority of cells in the human body) are withdrawn from the cell cycle in a state named G0 (G zero).
The p16-cyclin D-pRb Pathway
The retinoblastoma gene was isolated in 1986. It was the first tumor suppressor gene that was isolated based on knowledge of its chromosomal location: chromosome 13 band q14. Germ line mutations in the retinoblastoma gene, Rb, predispose to a pediatric malignancy of the eye: retinoblastoma. In addition, loss of Rb predisposes to a variety of other tumors later in life, with osteosarcoma being the most prominent secondary tumor. Loss of Rb function is also seen in a variety of spontaneous human tumors, including lung cancer, lymphoma and breast cancer.
The retinoblastoma gene encodes a 110 kDa phosphoprotein (pRb) that is expressed in almost every cell of the human body and contributes to growth regulation in these cells. Reintroduction of a functional Rb gene in retinoblastoma tumor cells results in growth arrest, indicating that the function of the gene is to restrict proliferation.
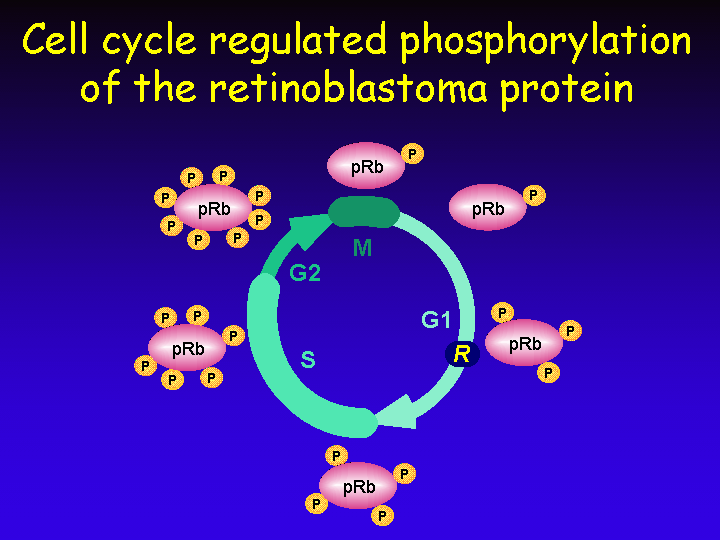
The retinoblastoma protein pRb undergoes cell cycle-regulated phosphorylation. In early G1 the protein is hypophosphorylated and its phosphorylation increases as cells progress through the cell cycle. During mitosis phosphates are removed and the cycle of phosphorylation starts over during the next G1 phase.
Phosphorylation of pRb is brought about by a group of related serine/threonine kinases named Cyclin-Dependent Kinases (CDKs). They are cyclin-dependent because their activity requires association with a regulatory subunit named “cyclin”.
In total some 15 CDK phosphorylation sites are present on pRb.
In mitosis a protein phosphatase 1-like protein (PP1) removes all phosphates from pRb to reset the phosphorylation status of pRb to the “hypophosphorylated” state of early G1.

The cyclins are named “cyclins” because their appearance during the cell cycle is cyclical.

The cyclical appearance of the cyclins is due only in part to a cell cycle regulated transcription of the genes that encode these cyclins. Their disappearance is mostly regulated through induced proteolysis.
Regulated proteolysis is a recurring theme in cell cycle regulation as protein breakdown gives directionality to the process: After a protein is destroyed by proteolysis, the cell cannot easily return to an earlier state in which the protein was still present. In contrast phosphorylation is easily reversible through the actions of phosphatases and is therefore not a good tool to assure that the cell cycle proceeds only in the forward direction.
An important question is how mitogenic signals that are received by cell surface receptors communicate to the nuclear cell cycle machinery to induce cell division.
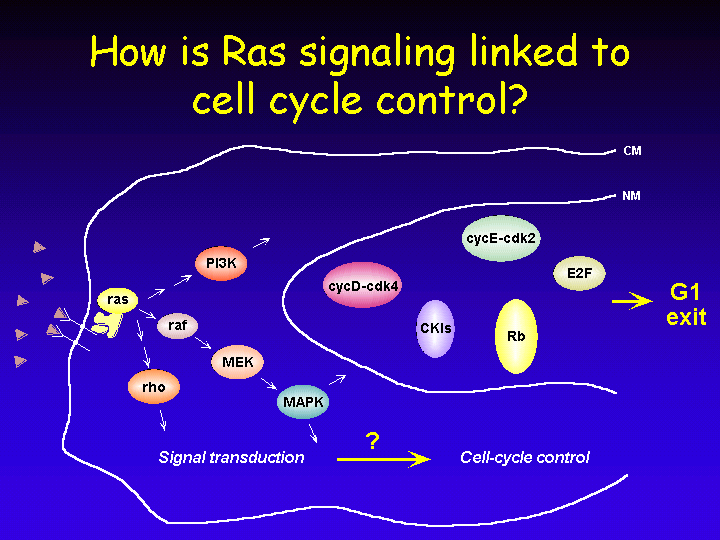
Ras is a frequent downstream target of growth factor receptors and in turn Ras signals to a number of cytoplasmic signaling cascades such as PI3-kinase, Raf and Rho. In their turn, these proteins connect to the nuclear cell cycle machinery to mediate exit from Go into G1 and S phase of the cell cycle.
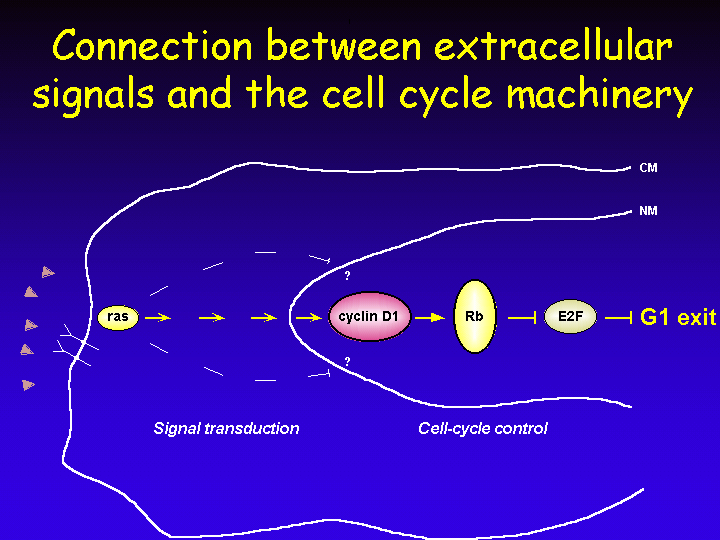
Expression of cyclin D1 occurs after mitogenic stimulation of resting cells. Expression of cyclin D is induced in response to a variety of mitogens and this has led to the notion that cyclin D acts as a “growth factor sensor” in the cell. Activation of Ras leads to transcriptional induction of cyclin D1 through a Ras-responsive element in the cyclin D1 gene promoter.
That cyclin D1 is an essential mediator of Ras signaling was demonstrated by using a dominant negative mutant of Ras: RasN17. Expression of this inhibitor of Ras signaling leads to G1 arrest, but can be rescued by ectopic expression of cyclin D1.
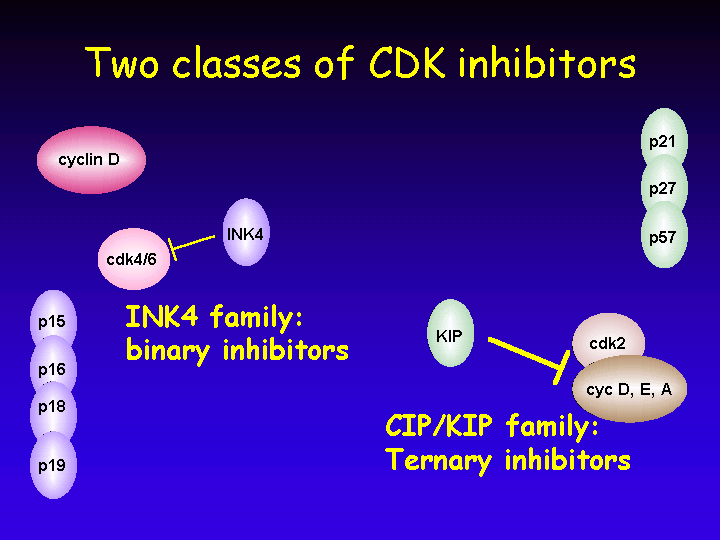
Another level of regulation of Cyclin-Dependent Kinases (CDKs) is exerted by CDK inhibitors (CKIs).
The inhibitors come in two flavors:
The gene encoding p16INK4A is a tumor suppressor gene: it is deleted in familial melanoma and also frequently mutated in spontaneous cancers such as pancreatic carcinoma.
Even though it was originally thought that this family of inhibitors were universal inhibitors of all cyclin/CDK complexes, it is now clear that they act as inhibitors of cyclin E/CDK and cyclin A/CDK complexes, but have the complete opposite effect on cyclin D/CDK complexes. For these complexes, the CIP/KIP proteins are essential assembly complexes in the absence of which cyclin D/CDK complexes cannot form.

The central role of CDKs in cell cycle regulation is probably best illustrated by the fact that many cell signaling pathways converge on these proteins.
As pointed out earlier:
In addition, CDKs undergo complex regulatory phosphorylations: phosphorylation of CDKs on specific threonine residues by CDK-Activating Kinase (CAK) is required for CDK activity, whereas dephosphorylation on specific serines and tyrosines by the CDC25 phosphatases is also required for activity of CDKs.
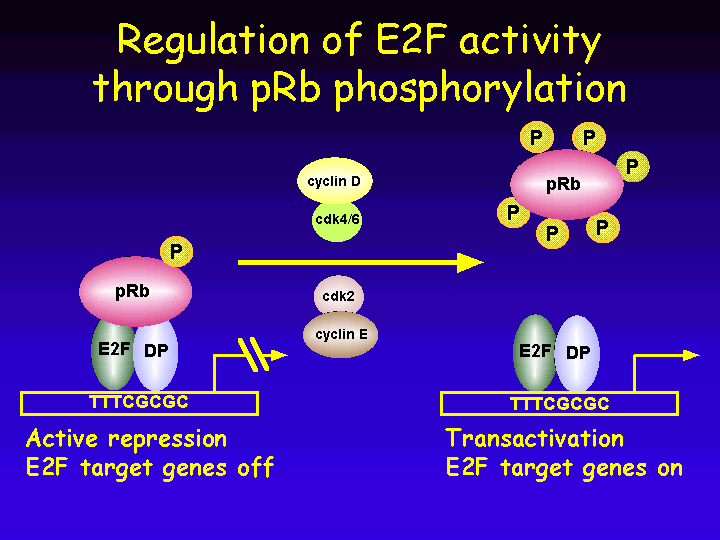
The most relevant target of the retinoblastoma protein is without doubt the cellular transcription factor E2F.
E2F activity consists of a heterodimeric complex of an E2F polypeptide and a DP protein. The E2F/DP heterodimeric complex can bind to specific DNA sequences in the promoters of E2F responsive genes.
E2F is a potent stimulator of cell cycle entry. Expression of E2F in resting cells is sufficient to induce entry into S phase.
Broadly, three classes of genes are under E2F control.
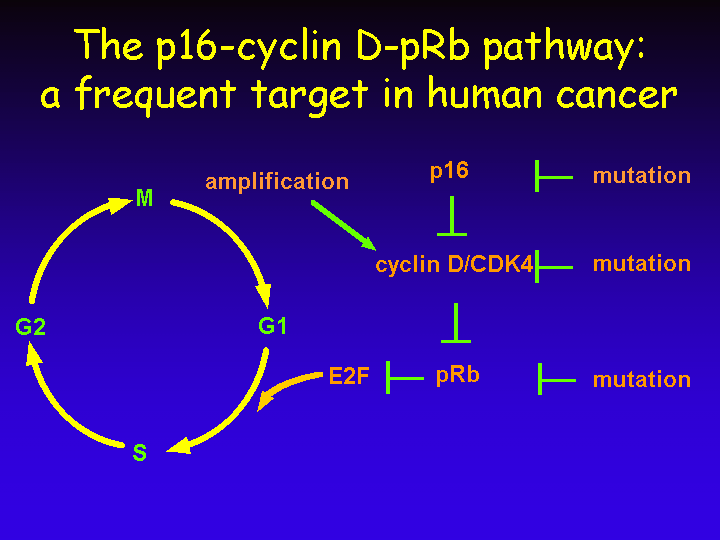
The central role of E2F in cell cycle regulation is probably best illustrated by the fact that many of the upstream regulators of E2F are mutated in human cancer.
Given the frequent mutations in each of these upstream regulators of E2F, it is now generally believed that virtually all forms of human cancer have deregulated this p16-cyclin D-pRb pathway in one or another way.
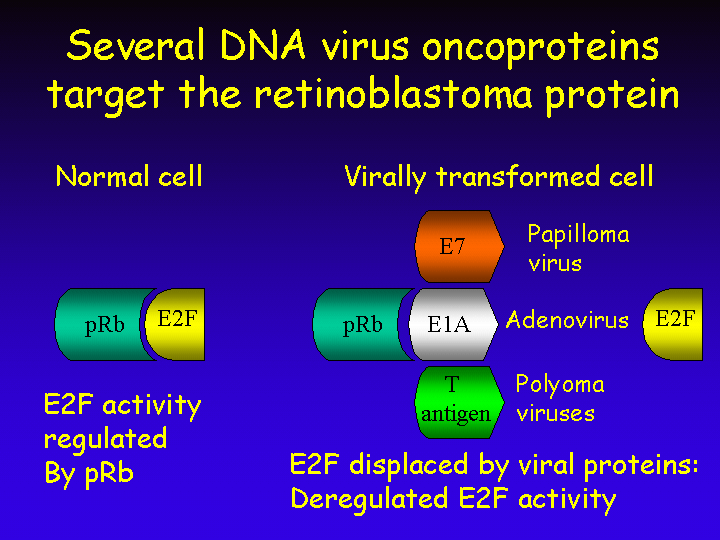
That pRb is central in cell cycle regulation is not a recent discovery of mankind. DNA tumor viruses knew this many million years ago and developed specialized viral proteins to bind and inactivate pRb.
The E1A protein of adenovirus for instance binds pRb with high affinity and thereby displaces E2F from the pRb complex. Similar proteins are encoded by human papilloma viruses (the E7 protein) and by polyoma viruses (their large T antigen).
These viral proteins were not primarily designed to transform infected cells but rather to induce S phase in the infected cells. These DNA tumor viruses do not encode their own enzymes for DNA replication and depend on the cellular DNA replication machinery to replicate their own DNA. Induction of S phase after infection of resting host cells is therefore essential for successful viral multiplication. Transformation by these viruses is merely a side-product of their ability to stimulate S phase entry.
The retinoblastoma gene has two relatives: p107 and p130. Together, the retinoblastoma protein family is called the “pocket protein” family and they are thought to have a binding pocket for cellular proteins such as E2F.
The genes encoding p107 and p130 are not genuine tumor suppressor genes as they are not found mutated in human cancers. Furthermore, inactivation of p107 or p130 in mice by homologous recombination does not predispose to tumor development. Nevertheless, over-expression of either p107 or p130 does induce G1 arrest in many human cell types.
The E2F family consists of five members:
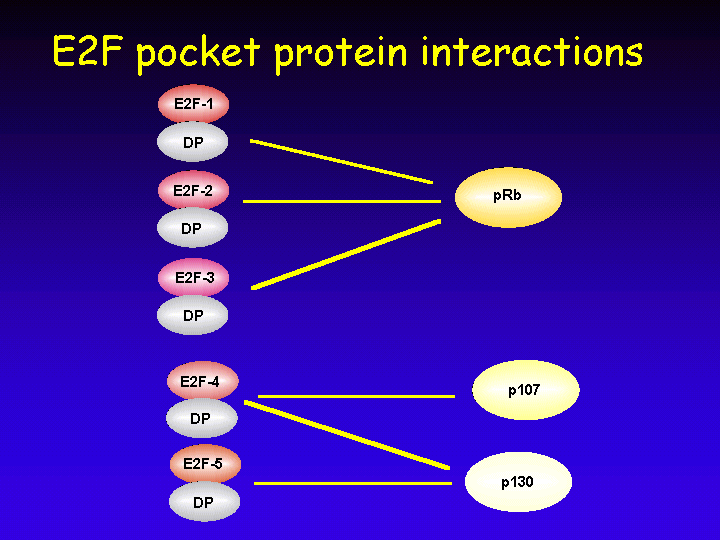

Some indications about what the different functions of the various E2F-pocket protein complexes could be stem from the analysis of the cell cycle-regulated appearance of these complexes. In resting (G0) cells, E2F-p130 complexes are most prominent as seen in this electrophoresis mobility shift assay (EMSA).
In contrast, exponentially growing cells have significantly more free E2F and also have E2F-p107 complexes.
As E2F-pocket protein complexes act to repress a family of proliferation-associated genes it has been postulated that the presence of E2F-p130 complexes in resting cells serves to suppress the expression of these growth-inducing genes to maintain the quiescent state. Therefore, E2F-pocket protein complexes can prevent cell cycle entry in resting cells.
In addition, there is now ample evidence to indicate that growth-inhibitory signals lead to an increase in E2F-pocket protein complexes, which in turn are responsible for down-regulation of these growth-stimulatory genes. As a result, E2F-pocket protein complexes are essential mediators of anti proliferative signals to force cell cycle exit.
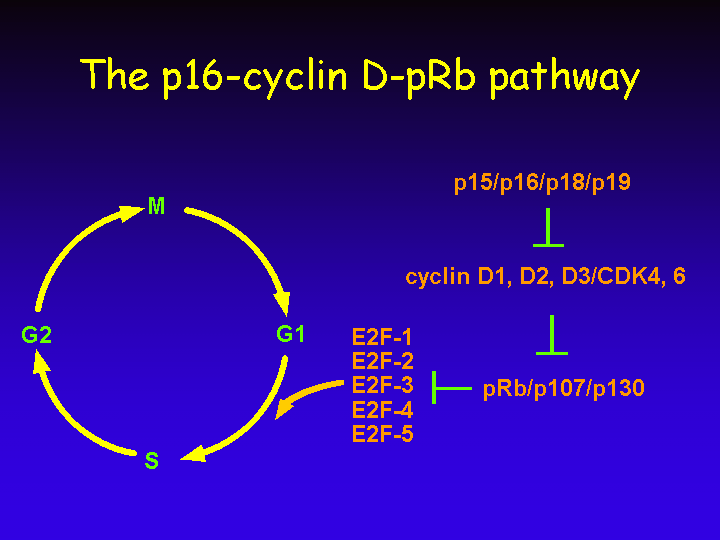
Most components of the p16-cyclin D-pRb pathway are redundant: they are encoded by a family of related genes.
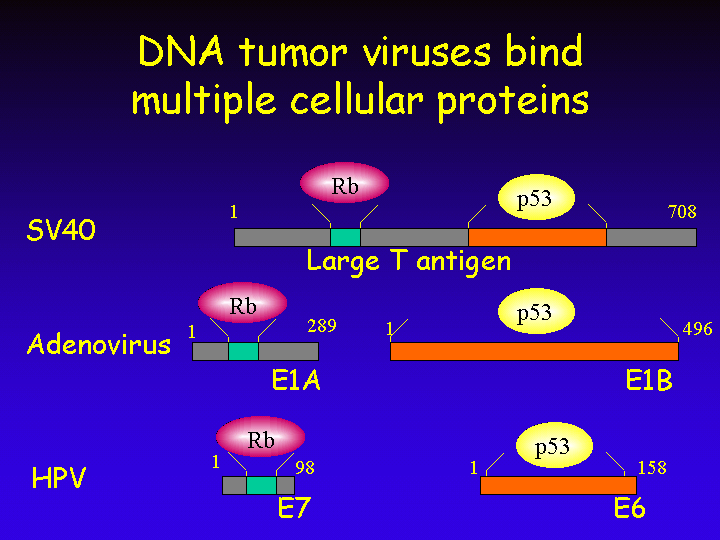
The transforming proteins of DNA tumor viruses not only target the retinoblastoma protein for inactivation, but also bind to another important tumor suppressor protein: p53.
The p19ARF - Mdm2 - p53 Pathway
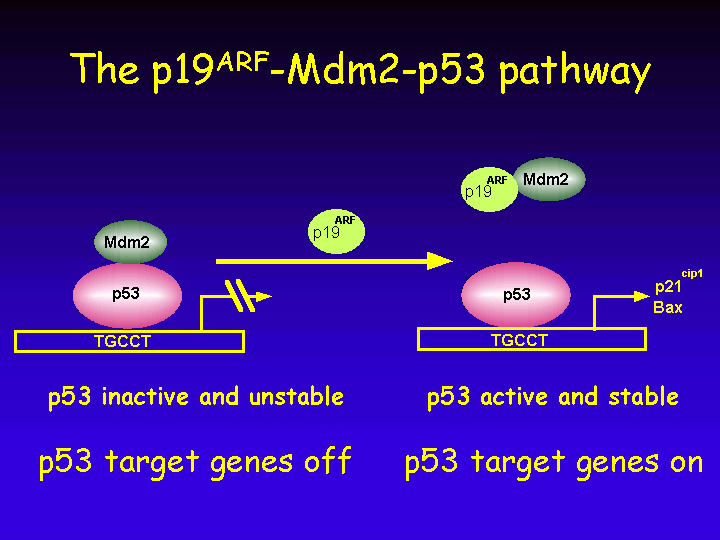
When cells are exposed to genotoxic agents, several checkpoints are triggered.

Similarly, when chromosomes are damaged as a result of genotoxic stress, cells will not enter mitosis until the damaged chromosomes are repaired.
For this reason, p53 is also called “the guardian of the genome” as the integrity of our genome is rapidly lost when p53 is mutated.
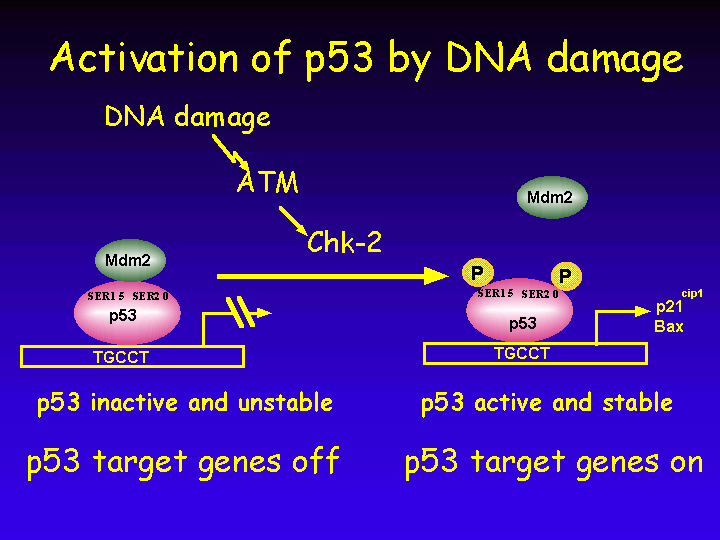
Activation of p53 after exposure to DNA damage is mediated by a cascade of protein kinases.
Thus, DNA damage leads to stabilization and activation of p53 and leads to a transcriptional induction of p53 target genes.
The INK4A Locus
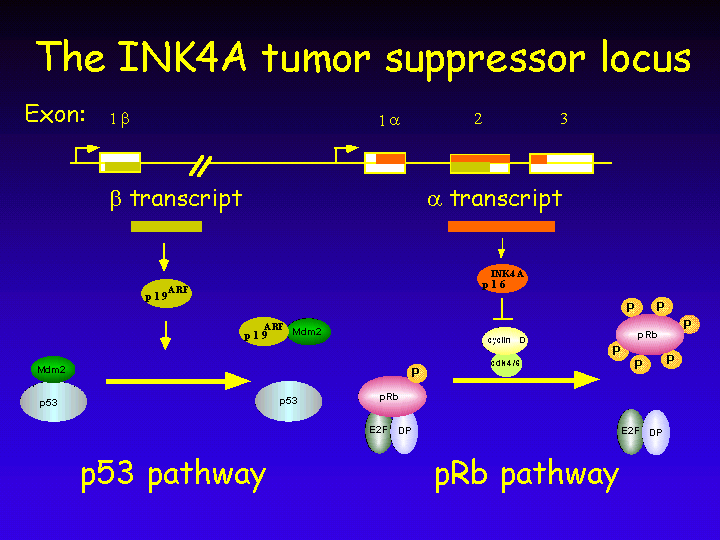
The INK4A tumor suppressor locus is highly unusual in that it encodes two completely different proteins from overlapping reading frames.
Thus, the INK4A locus encodes tow proteins that act on the p53 and pRb pathways, respectively. At first glance it would appear undesired from an evolutionary perspective to have two central pathways in growth control regulated by a singly genetic locus. After all, mutation in one locus inactivates two growth-inhibitory pathways, making a species that has such a locus more cancer-prone. The advantages of having this genomic organization are discussed below.
The p53 and pRb pathways of growth control are not separate but highly interconnected.
Binding sites for E2F are present in the promoter of p19ARF. As a result, inactivation of pRb leads to activation of p53 in the cell.
Furthermore, Mdm2 does not only interact with p53 to negatively regulate it, Mdm2 also binds and inactivates pRb.
Recent work from our own Institute indicates that loss of retinoblastoma-family function leads to insensitivity to growth-inhibitory signaling through the p19ARF-p53 pathway.
As is discussed below, both signaling through Ras and the polycomb protein Bmi1 leads to upregulation of expression of both genes in the INK4A locus, causing both a p53- and a pRb-dependent response.
A cell has to make sure that a single mitogenic stimulus does not lead to multiple rounds of DNA replication. For this reason, negative feedback loops exist in the cell to limit cell division in response to mitogens. One such negative feedback loop is shown here.
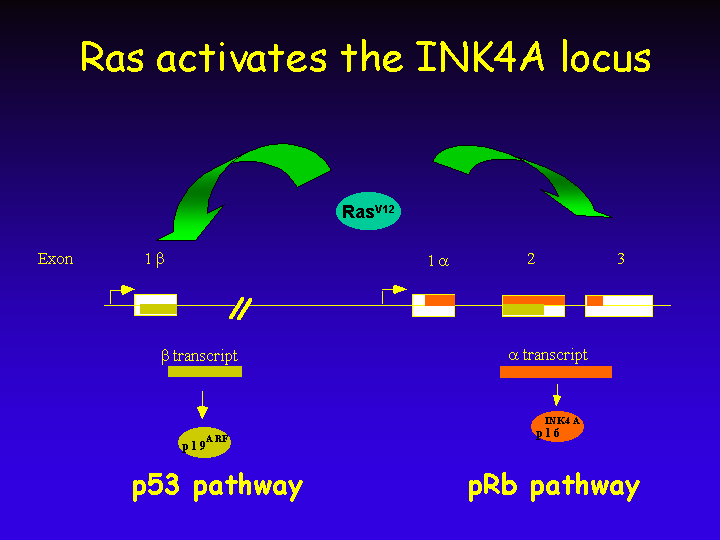
This transcriptional response activates both the growth-inhibitory pRb and p53 pathways, thereby limiting the cellular proliferative response to mitogens.
Ras-induced Senescence
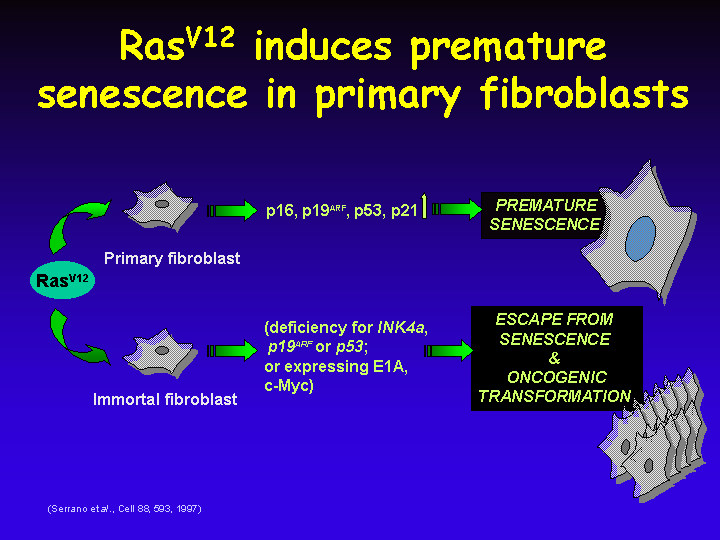
It is now clear that loss of the p53 pathway is most relevant to allow escape from Ras-induced senescence, as cells that lack either p53 or only p19ARF also become transformed by a Ras oncogene.
In addition, expression of genes such as c-myc or adenovirus E1A allow escape from Ras-induced senescence.
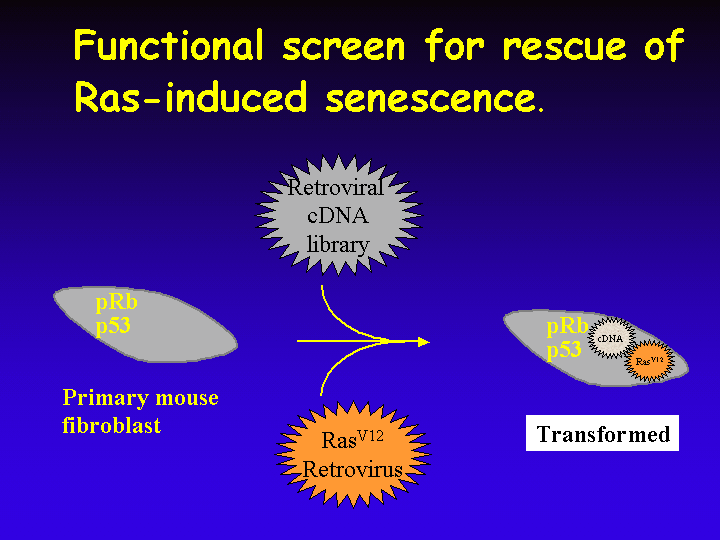
In my own laboratory we are searching for genes that allow primary cells to escape from Ras-induced senescence. To do this, primary mouse fibroblasts are infected with a retroviral cDNA expression library and a virus that encodes oncogenic Ras.
Rare cells that undergo transformation rather than premature senescence can be readily identified in a senescent population.