Oncogenes:
proliferative or apoptotic response?
by G.
Evan
Cancer
Cancer is a very common disease, affecting about 1 in 3 individuals, and about half the people that contract cancer will die as a direct result of their disease.
For the most part, cancer arises from a single cell, that is, cancer is a clonal disease. The average human being contains about 1014 cells (i.e., 100,000,000,000,000 cells), any one of which could, in principle, become a cancer cell, if it acquired the right sort of mutations while it still had the potential to proliferate. Therefore, the cancer cell arises and progresses once out of a possible 1014 cellular targets. That only happens in 1 in 3 people. Even then it usually takes 60 or 70 years to occur.
Why is the cancer cell so rare?
This is a profound problem for multicellular organism: how can cells proliferate and survive under normal conditions, but not to become neoplastic following the acquisition of mutations. The usual current explanation for the rarity of cancer is the argument that cancer is a multistage disease that requires multiple independent mutations in order to convert a normal cell into a tumor cell.
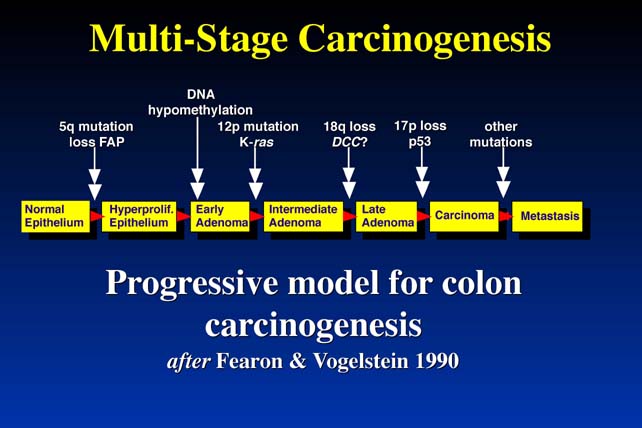
This is the diagram depicted a decade ago by Fearon and Vogelstein to demonstrate the multistage nature of carcinogenesis in the colon. A normal colonic enterocyte undergoes a mutation in a gene that regulates cell growth, and begins to expand clonally. Initially, the expansion is very indolent, because the development of a malignancy requires not one mutation, but maybe up to 12 or more independent mutations.
The chances of
any one mutation occurring, amongst all the billions of cells in the gut, is
quite high in a life-time. But the chance of two mutations occurring in one
cell is, of course, the square of that probability, and so on. In fact the chance
of all twelve mutations occurring in one cell in the lifetime of an individual
is very small.
Problems with the current model
This model is - in general terms - correct. Cancer is a multistage disease. But there are two very serious problems with this explanation of the rarity of cancer.
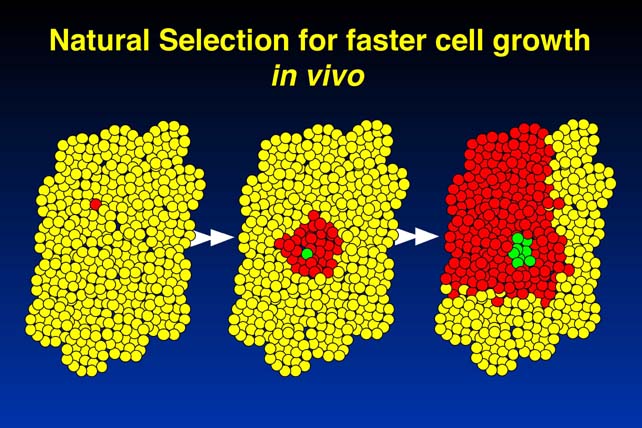
The first problem is that it does not take into account the fact that mutations that give rise to cancer cause clonal expansion. After the first cell undergoes a mutation, it begins to expand clonally. Let's say it expands to 108 progeny cells. This would correspond to about 20 - 25 cell divisions, and 108 cells is a very, very small nodule. The problem we now have is, we have to look at the possibility of mutations occurring in anyone of those 108 progeny cells. Any mutation that makes one of those progeny cells grow even more quickly leads to further clonal expansion. This will therefore increase the chance that one of those progeny get another mutation, and essentially the first mutation drives the second, drives the third, and the system drives itself into malignancy. This is a very serious problem, and correspondingly, the rarity of cancer cannot be explained by the need for multiple mutations.
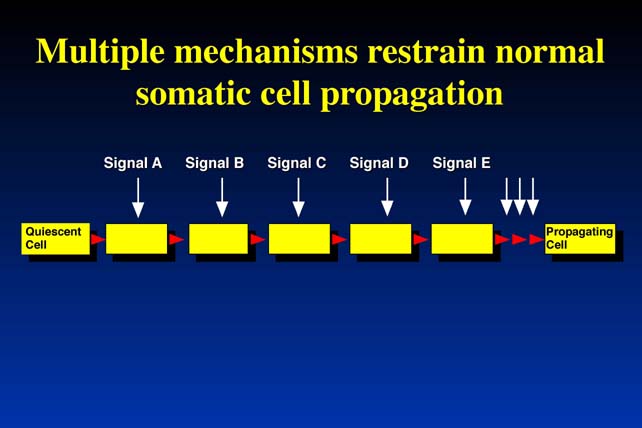
The second problem comes when we try to consider not what separates a normal cell from a tumor cell, but what separates a quiescent cell (i.e., that is not proliferating) is resting - from a cell of the same type that is normal, but is proliferating and surviving. It follows that, if there are independent mechanisms, which prevent normal cells from becoming tumor cells, that is from proliferating uncontrollably, then every time a normal cell needs to proliferate these independent mechanisms have to be turned off. Because they are independent each one needs a different signal to turn it off. The problem can be stated in simple terms:
Potential solution
There actually is a very simple solution to this biological dilemma. The mutations that arise during the progression and development of cancer cannot occur in isolation. The requirement for clusters of mutations, each which rely upon the other, makes the development of cancer very rare. The following sections offer an explanation of this phenomenon.
The c-myc Oncogene
|
The
c-myc Oncogene Cellular
homologue of viral v-myc oncogene of avian myelocytomatosis virus. Several homologues in man:
c-myc
is expressed in differentiated proliferating cells c-myc
is repressed during differentiation and growth arrest |
I would like to illustrate one potential explanation for this by looking at the c-myc oncogene. c-myc is the cellular homologue of a retroviral oncogene, first defined in a group of retroviruses that cause myelocytic leukemia in chickens. Like almost all retroviral oncogenes, the viral myc gene (v-myc) is derived from a cellular progenitor (c-myc).
1. myc induces proliferation
c-myc is expressed in proliferating cells in the body, such as keratinocytes, hepatocytes, bone marrow cells, fibroblasts, and vascular smooth muscle cells. It is repressed when these cells withdraw from the cell cycle.
myc deregulation
is associated with various types of cancer. myc becomes deregulated by a variety
of different mechanisms:
Deregulated c-myc causes proliferation but not expansion
myc is an oncogene because deregulated myc is sufficient to put cells into cycle and to keep them there. This is potentially dangerous, because any myc mutation amongst the billions of cells in the body could deregulate cell proliferation checks and lead to cancer. However, this is a relatively rare event: why is this so? The explanation seems to be obvious when you look at how these cells grow in culture.
This figure shows fibroblasts transfected with constructs containing activated c-myc (Rat-1/c-Myc) compared to control (Rat 1/Neo). Live cell number is measured over time.
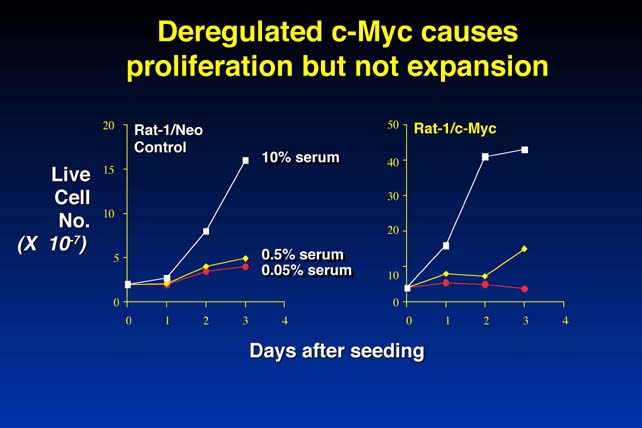
There is no increase in cell number because, as well as inducing proliferation, in low serum myc induces apoptosis ("cell suicide", or programmed cell death). The cells degrade their DNA, bleb and fragment, the DNA condenses, the cell fragments are phagocytized by the neighboring cells. Therefore, myc is not only a potent inducer of proliferation, it is also - under certain circumstances - a very potent inducer of the completely opposite biological program, cell suicide or apoptosis.
The proliferation - apoptosis coupling
This is true not only for myc; every mechanism by which growth can be deregulated in mammalian cells not only induces proliferation, also induces - under certain circumstances - apoptosis. Some examples of this are:
All can induce proliferation and apoptosis. This observation provides us with a potential hypothesis for the rarity of cancer:
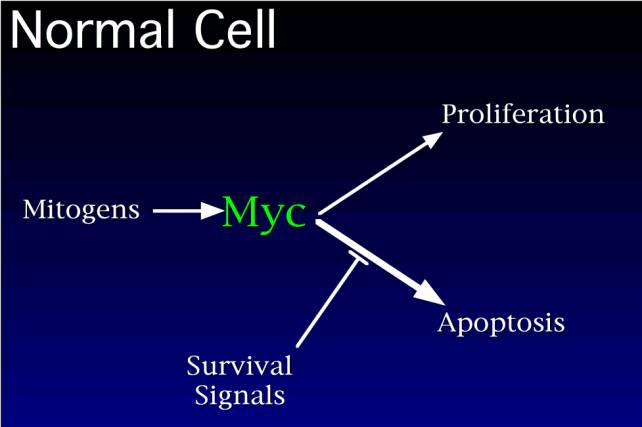
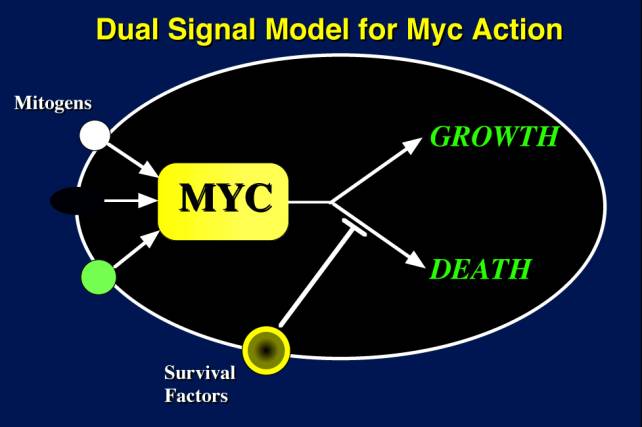
In normal cells, mitogens activate myc and the proliferative machinery. myc then implements two pathways: a proliferation pathway and a death pathway. The reason cells don't all undergo apoptosis is that the apoptosis pathway is blocked specifically by other survival factors that are provided by the cell's neighbors. So when a cell is triggered into the cell cycle in the correct environment where it gets the right survival factors from its neighbors, the apoptotic program is suppressed and cellular proliferation follows.
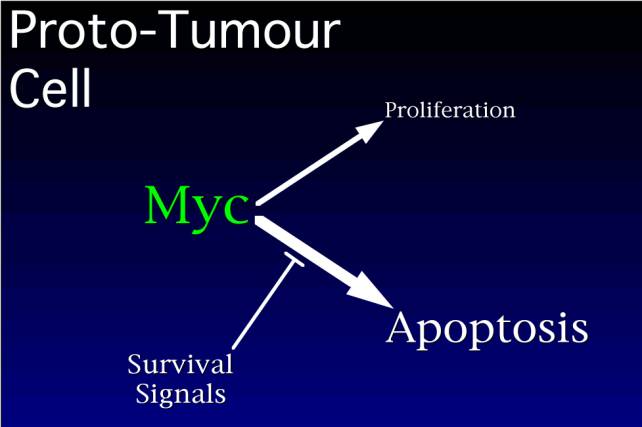
Let us imagine that myc becomes activated by a mutation. At high levels of myc, proliferation is superactivated, and apoptosis is superactivated. The cell begins to proliferate, but it and its clonal progeny exhaust the supply of survival factors, and apoptosis begins to predominate, and the very mutation that is making these cells proliferate, now kills them off.
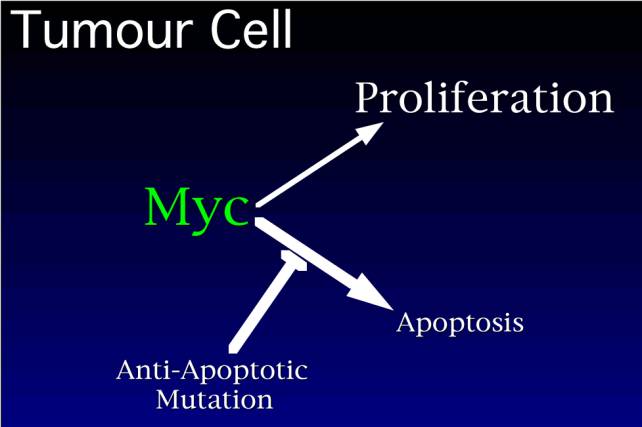
How then do cancers
arise? The idea would be that cancers can only arise if they acquire an anti-apoptotic
mutation, which blocks this pathway, leading to net proliferation. Is there
any evidence for this?
The oncogene Bcl-2
The Bcl-2 oncogene is activated in follicular B-cell lymphoma in human beings. Bcl-2 is on chromosome 18 and gets activated by chromosomal translocation to the immunoglobulin gene locus- t(14;18).
Bcl-2 is an oncogene not because it makes cells proliferate, but because it blocks apoptosis. You can see therefore that there is a possible interaction between Bcl-2 and myc. myc drives proliferation and apoptosis, but Bcl-2 blocks apoptosis, which results in uncontrolled proliferation.

This figure shows fibroblast under low-serum conditions, in which myc is under the control of a molecular switch. At the upper left, c-myc is off, and fibroblasts are quiescent. On the upper right, when c-myc is turned on, the cells go into cell cycle but undergo apoptosis and the culture dies. At the bottom right, we see the situation with co-expression of myc and Bcl2: apoptosis is blocked, and cells continue to proliferate.
Oncogene Cooperation
The paradigm for oncogene cooperation comes from classic experiments by Land et al. some 15 years ago:
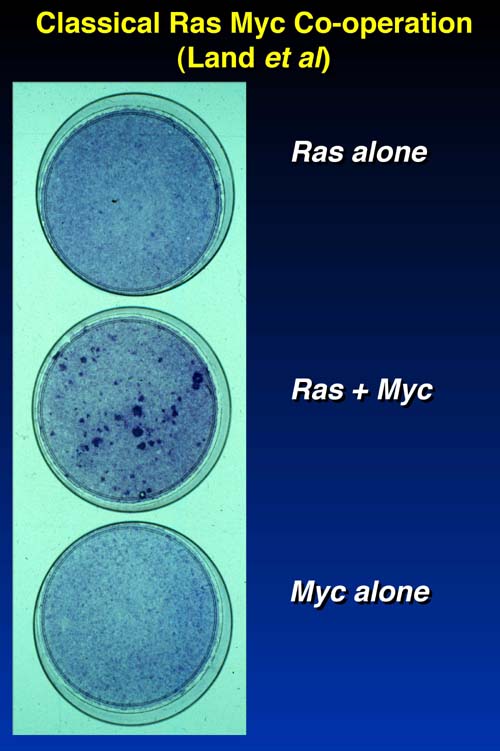
Fibroblasts with activated ras (top) or activated myc (bottom) alone do not undergo transformation. However, co-expression of activated ras and myc (middle) does lead to foci of transformed cells.
If myc and Bcl-2 are co-expressed in similar experiments, cells survive, but foci of transformed cells are not observed. This suggests that the way myc and Bcl-2 cooperate is different from the way that ras and myc cooperate.
How do ras and myc cooperate to form tumors?
Experiments done about 10 years ago with transgenic mice where activated ras, myc, or both are expressed in mammary tissue. Tumor-free status was examined over time.
We can ask a simple question: does ras cooperate with myc because it blocks myc-induced apoptosis? Experiments with fibroblasts showed that the answer to this question is no. Activated ras increases cell death in fibroblasts also harboring activated myc. We might conclude that ras plays no role in the suppression of myc-induced apoptosis. However, this conclusion is totally wrong. The reason it is wrong is that ras does indeed suppress apoptosis, but at the same time it promotes apoptosis because ras has two different effectors.
Importantly, oncogenes
and their protein products often have more than one function, and the effects
of a combination of oncogenes are often different from their effects in isolation.
The combination of ras and myc, or of myc and Bcl-2
may have completely different effects than ras, myc, or Bcl-2
alone. Also, ras may cooperate differently with myc than with
Bcl-2.
Attributes of the cancer cell
Attributes of cancer cells that are associated with the development of tumors were elegantly reviewed by Hanahan and Weinberg in Cell.
In summary:
One could think about oncogenes and cooperating mutations in cancer, in terms of mutations that eliminate each of these growth inhibitory functions.
The role of survival factors
If the decision whether cells grow or die following myc activation depends on survival factors, then we need to know what the availability of survival factors is in living tissues. The problem is we do not really know what the survival factors are, and we certainly do not know what their concentration is because in many cases survival signals come from direct cell-cell contacts. We do not know whether survival factors are in excess or in limiting supply.
Let us do a thought experiment:
If you were thinking about a way of treating that tumor, then putting back into those tumor cells the apoptotic program they have lost, would be a very effective and specific way of curing that disease.
Conclusions
I have tried to illustrate that we can learn a lot about tumorigenesis from transgenic mice and by manipulating various attributes of carcinogenesis. However the way that these events may occur will differ from tissue to tissue. The twin properties of deregulated proliferation and suppressed apoptosis are fundamental to understanding how cancers arise and emerge in human beings. These are two, but only two, of the critical pathways that we would like to be able to influence with new drugs. It does not exclude the fact that other targets are also important: angiogenesis, invasion and immortalization, but it does show that the suppression of apoptosis is fundamental to the emergence of cancer in all tissues in the body.