P53
- an emergency brake for growth control at the interface between selection pressure
and growth control
by Pierre
Hainaut
In the following, I will talk about p53 and its relationship to cancer, and will primarily address three questions:
Many lectures in
this course have focused on cancer pathology (shapes and patterns), cancer genetics
(sequential alterations in genes and gene expression), and the molecular biology
of cancer (microchips and computer network signaling pathways).
However, one very important point has not yet been covered sufficiently: the
cell's role in cancer.
The development of cancer requires a combination of several types of events: cells must acquire the ability to grow autonomously, increased genetic instability develops, cells develop an unlimited replicative potential, and there are alterations in angiogenesis and invasiveness.
Selective Pressure
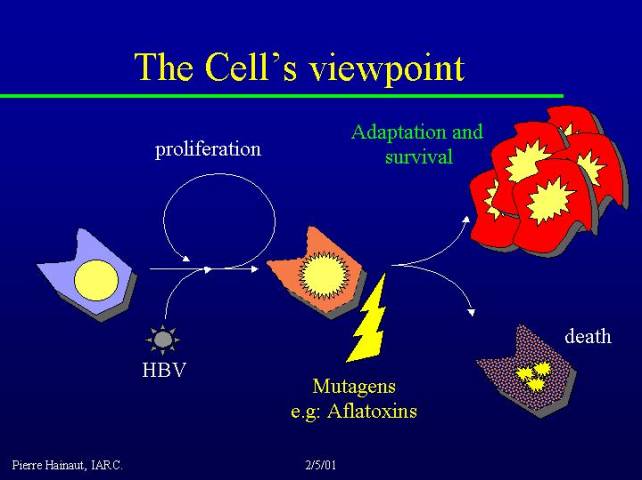
There are several pathways leading to the development of cancer, but it is very important to realize that they are all very dependent on selective pressure. Let us imagine a hepatocyte in the liver of a 40 - 50 year individual who is a chronic carrier of the hepatitis B virus (this is very common, for instance, in Senegal). The chronic carrier state leads to chronic cellular stress and damage.
Cells experience oxidative stress, neighboring cells secrete cytokines, and the situation becomes even worse, so that the cell in our example is likely to die. To compensate for loss of cells, the liver increases its capacity to proliferate: this is a very simplified and schematic representation of the condition known as liver cirrhosis.
If there are mutagens in the environment (for instance, the mutagen aflatoxin is very common in the diet of individuals living in Senegal), the cell has an increased risk of developing mutations. One can imagine a Darwinian context, in which individual cells experience selection pressure as to which of the mutated individual cells are particularly fit to survive in this perturbed environment. This is what will lead to become a cancer: under this kind of selection pressure in the liver, the cell's choice is to die or to try to adapt by becoming a cancer cell.
p53: interface between selection pressure and growth control
A very important component of cancer is the interface between this environmental selection pressure and the growth control machinery. This is the role of the p53 protein.
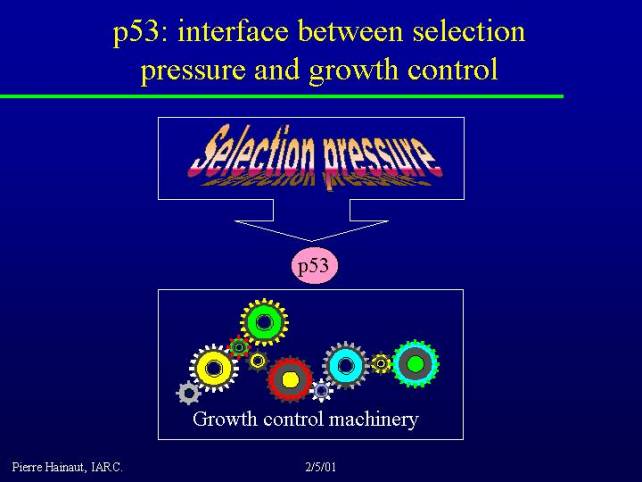
The p53 pathway lies at the interface between all the many signals that can come from the environment and the control of the proliferative machinery.
p53 was first identified
about 21 years ago.
It took about 10 years to realize that it was a tumor suppressor.
The first p53 mutations were identified in cancer (losses of alleles) in 1989.
10 years later (1999) the number of mutations described in cancer was 10.000.
We now have over 14.000 mutations gathered in the database
that we maintain at the IARC
p53 as a sensor of multiple forms of "stress"
The activity of p53 is due to a number of biochemical characteristics. The first is that the protein is inducible by multiple forms of stress. Not only by DNA damage but also by other forms of stress which are not genotoxic in themselves. For example senescence has been illustrated as one possible mechanism that leads to p53 accumulation.
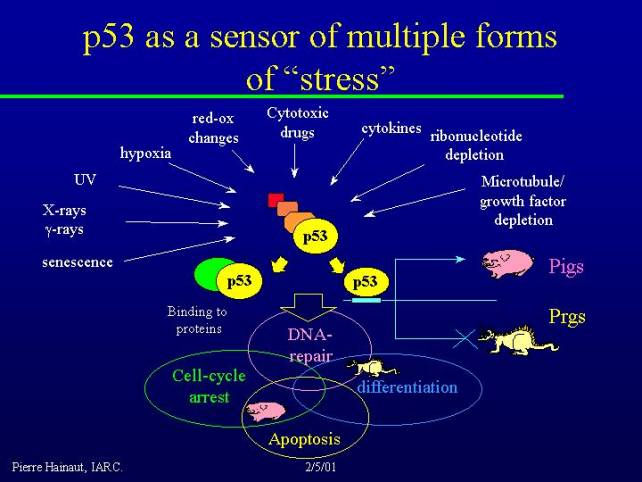
The protein is present in normal cells and is expressed at a relatively constant level, but it does not accumulate and it is also in an inactive conformation. The process of activation requires changes in the conformation of the molecule and accumulation of p53. Therefore, both qualitative and quantitative processes result in p53 activation. This can happen due to a variety of processes. For example:
p53 is thus a very broad sensor of many stress conditions. Activation of p53 can cause it to bind to other proteins, as well as to act as a transcription factor and induce the transcription of genes which are now collectively called PIG's for p53-induced genes. Quite recently there has been an emphasis to on the notion that p53 also represses the transcription of genes, so there are also p53 repressed genes, PRGs.
PIG's and PRG's are regulated by p53, and all together PIG's and PRG's regulate a set of integrated processes:
Anatomy of p53
P53 is a relatively small protein: 393 amino acids, with the typical structure of an transcription factor:

Mutations in the p53 coding sequence
It is interesting to note that most known mutations in cancer affect the DNA binding domain.

This slide is based
on the 10.000 mutations from the database 1999.
As you can
see 90% of them are located in the central DNA binding domain.
A very striking point is that most (70%) of these mutations are missense mutations. This is at variance with many tumor suppressor genes where the alteration has as an effect to truncate the protein or to remove it altogether because there are insertions, deletions or frameshift mutations.
p53 missense mutations lead to the stabilization and the presence in the cancer cells of a mutant protein, which remains there and is even selected in cancer, because you can find it in for example in distant metastases.
In some way, cancer cells seem to like mutant p53 and to retain it. That is probably due to the fact that this mutant p53 has some sort of (as yet unknown) function that may participate in a positive manner in the tumorigenic process.
Another very striking point if you look at the distribution of these mutations is that the N and C terminal regions carry very few mutations. 27of the approximately 14000 described mutations are in the extreme N and C terminus. Yet these regions contain very important regulatory signals: the phosphorylation sites, the acetylation sites that are important for the control of p53 functions; yet they are not targeted by mutagenic events in cancer. So it is possible that mutant p53 in cancer retains these intact N and C terminal regions because it is useful for some biological processes that still remain to be identified.

A diagram of p53 based on its partial crystal structure.
This diagram represents the crystal structure of the p53 protein.
The DNA binding domain is by far the most common site of mutations, and forms a semi rigid fold made of a scaffold of beta sheets, loops and alpha helices that together bind in the minor and major groove of DNA. This diagram shows the three amino acid residues that are the most frequently mutated in all cancers (they represent about 20% of all mutations in cancer):
This illustrates that the common p53 mutations have two main primary effects:
The P53 family
It was discovered about 3 years ago that there are other proteins with the same structure and similar biological properties with p53. Two of them have been identified: p73 and p63.
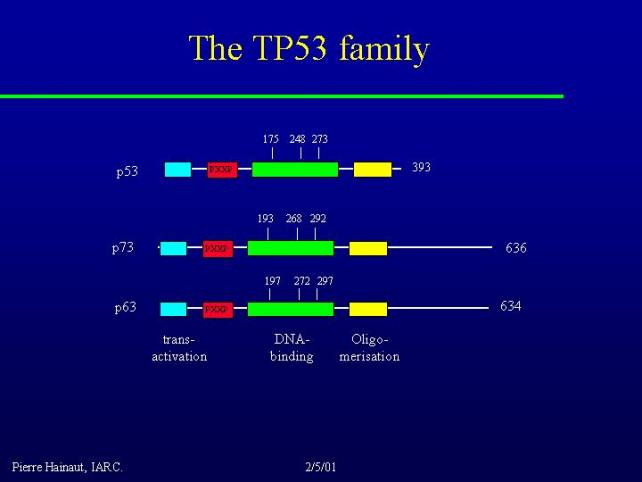
p63 and p73 have the same DNA binding domain with conserved residues corresponding to the most frequently mutated residues in cancer. They have also N-terminal transactivation and C-terminal oligomerization domains.
They can not only form oligomers with themselves but they can form hetero-oligomers with the other members of the family.
This is even more complex than this because p73 and p63 have two different variants.
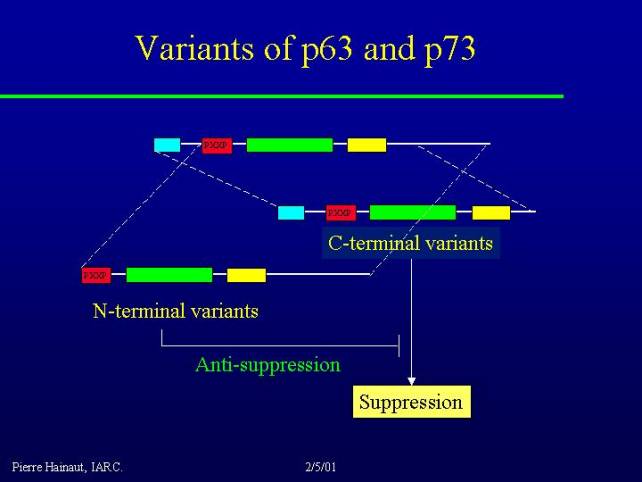
p73 has at least two major C-terminal variants, alpha and beta. These forms of the protein have probably mostly suppressor activity through DNA binding to the same kind of target than p53 and regulation of their activity.
There are also N-terminal variants, that means protein that have lost the N-terminal transcriptional activation domain. These proteins are not able to transactivate gene expression on their own, however they still interact with those and exert a sort of antisuppressive effect, by blocking their capacity to activate gene expression. Thus, there are complex regulations and interactions between the various members of the p63 and p73 family.
I would like to come back to what I said about the properties of mutant p53 in promoting cancer. Because mutant p53 conserves an intact C-terminus and retains the capacity to interact with some of the members of the p63 and p73 family, blocking and modifying the effect of these proteins might be one of the mechanisms by which mutant p53 may promote cancer.
Activation of p53 by stress signals
I will now briefly discuss how p53 senses different kind of signals, and how it becomes activated by stress.
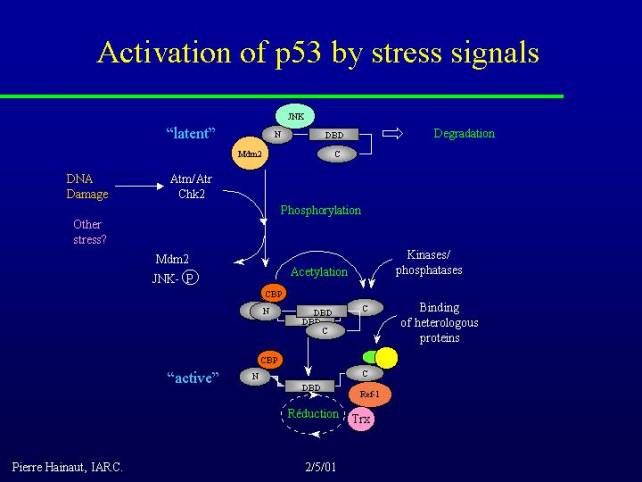
This figure is the representation of p53 in what I would call the latent situation. The molecule has a very particular conformation where the C-terminus is folded on to the DNA binding domain preventing it from recognizing target DNA. In addition the protein binds in its C-terminal region with at least two distinct proteins, one is called MDM2 and the other is the inactive form of the jun-kinase (JNK). These two proteins mediate the targeting of p53 to the proteasome for rapid degradation. So not only can the protein not bind DNA but also it is very unstable, so it disappears very rapidly from the cell.
Various stimuli are capable of releasing these interactions, of course DNA damage, through a pathway which has now been partially elucidated involving genes like ATM or its close homolog ATR and the cell-cycle regulatory kinase Chk-2. These various kinases are capable of phosphorylating p53 in the N-terminus and of releasing the interaction with MDM2. Activation of the jun-kinase too, maybe by a different pathway, leads to its dissociation from p53.
In this form p53 becomes capable of binding another family of proteins: CBP or P300. These proteins have histone acetyl transferase activities, and are coactivators of many transcription factors. In the case of p53, this histone acetyl transferase activity can exert itself on the C-terminus of the molecule and release the negative interaction of the C-terminus with the DNA binding domain. As a result the protein changes conformation and exposes its DNA binding domain.
These changes stabilize by a number of phosphorylation events that affect the C-terminus of the molecule and the final step in the process of activation consists in the binding of heterologous proteins in the C-terminus as well as in a step of oxidation-reduction because there are crucial cysteine residues in the DNA binding domain of the protein, which needs to be correctly reduced for the protein to have a maximum affinity for DNA.
A variety of factors may play a role in that such as the REF1 enzyme which is a double redox enzyme and DNA repair activity as well as thioredoxin.
This shows several ways, several levels at which p53 is capable of sensing things that happen in the cellular environment:
Therefore, p53 has the profile of a very complex sensor of all these different forms of stress. Once it is activated, p53 plays multiple roles and what I want to stress is that this roles happen at many different levels.
p53 and the cell cycle
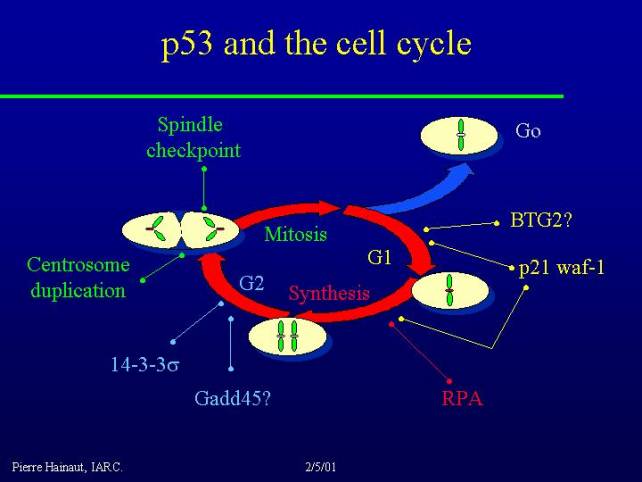
For example in cell cycle it is clear that p53 activates a protein like p21 WAF and this leads to cell cycle arrest in G1 and probably also in the G2/M interface.
There are also many other targets of p53 activity that play a role in the regulation of cell cycle. Importantly, p53 does not have a single, unique role, but possesses many, many different functions. Loss of p53 function does not alter a unique cell cycle checkpoint, but acts at many, many different levels. The same holds true for the role of p53 in apoptosis.
p53 and apoptosis
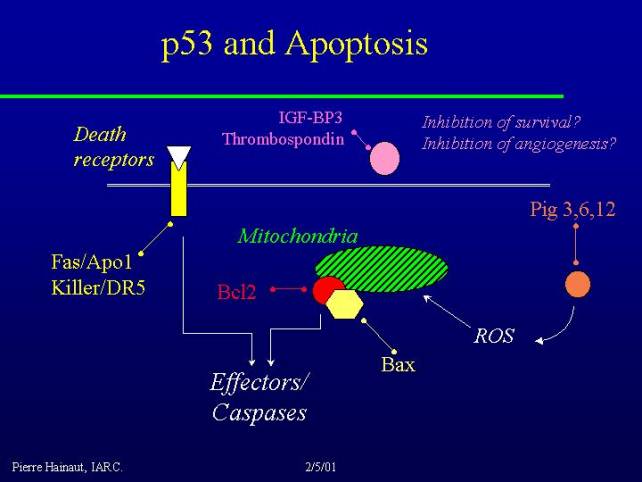
There are different ways to induce a cell to die, and one of the classical ways is to stimulate death receptors, such as for example the TNF alpha receptor, while p53 regulates the expression of at least two of these receptors FAS/APO1 as well as KILLER/DR5 and activation of these receptors can directly trigger a cascade of effector caspases leading to apoptosis.
Another classical route of induction of apoptosis is the alteration of the mitochondrial membrane and leakage of mitochondrial component which also activate effector caspases, and p53 can regulate this through the production of reactive oxygen species by families of genes which are now described as PIG3, PIG6, and PIG12 - these are all oxidoreductases involved in the production or control of reactive oxygen species with, as a result, mitochondrial damage and stimulation of an apoptotic pathway.
A classical target of p53 is the BAX gene, which is induced by p53, and BCL2 which is downregulated by p53. These two proteins play a role in the control of the mitochondrial membrane, so it's another way p53 can play a role in the regulation of apoptosis.
p53 is also involved in the control of the activity of several factors that may play a role as survival factors, including IGF.
Activation or regulation of survival factors may change the level of factors that are available to stimulate the survival or to maintain the survival of cells, and modifying of these molecules could also be a pathway by which p53 control apoptosis.
P53 mutation and tumorigenesis
I will try to explain why p53 is so frequently mutated in cancer. There are three main hypotheses, which are nit mutually exclusive:
Of course these three hypotheses are not mutually exclusive.
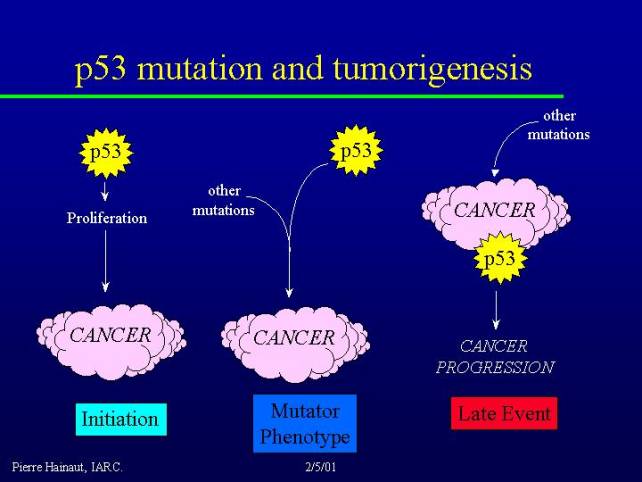
P53 mutations and the causes of cancer
We have 14.000 different mutations reported, corresponding to about 450 distinct sequence changes. That means that some mutations occur very frequently although some of them are rare. They differ by the chemical nature, by their position into the p53 gene; this allowed making tumor mutation spectra. The mutation spectra show variations with tumor types also variation with the geographic origin of the patients as well as with the tumor risk factors, providing clues on the mechanisms of mutagenesis.
It is very easy to classify the mutations in main categories according to what is expected to be the nature of the mutagenic event. There are two main types of mutations:
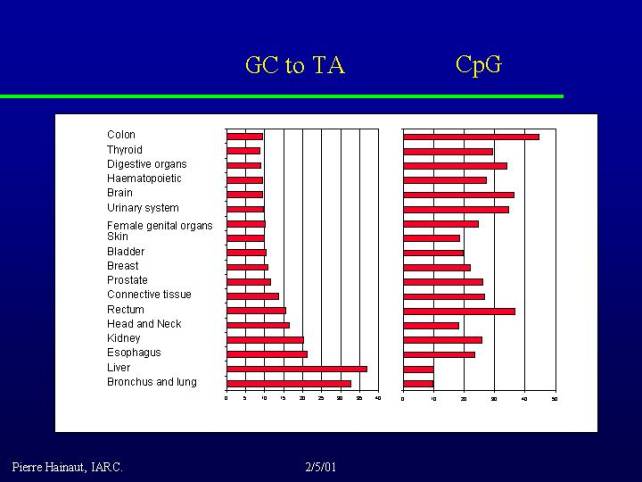
At the bottom of the graph in particular you see a number of cancers such as head and neck tumors, cancers of the esophagus, liver and of course bronchus and lung - where tobacco has the major etiology. These cancers have a lot of transversions and few transitions. So there is a correlation between the presence of transversions and the number of environmental exposures to serious carcinogens.
The pathways for induction of C to T mutations:
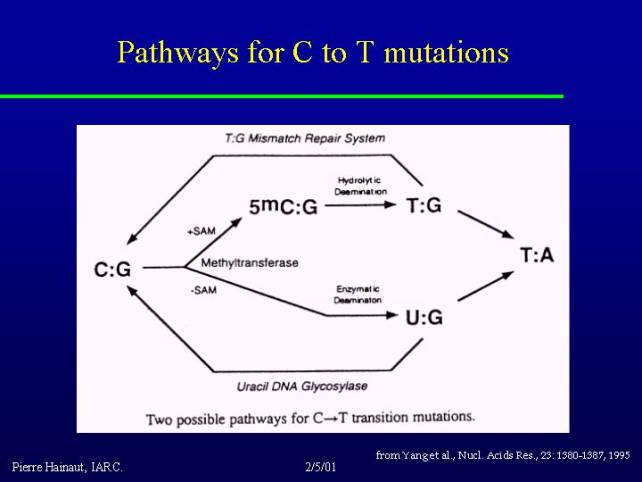
There are several possible pathways but a very common pathway is the consequence of the fact that about 3 to 5% of all the cytosines at CpG motives in the genome are methylated. 5-methyl cytosine can spontaneously deaminate to form a TG mismatch. If this mismatch is not repaired, it becomes a mutation. This happens very commonly in the whole genome at an important frequency.
I show you some examples of different distribution of mutations in a number of tumors along the digestive tract including two different types of cancer in the esophagus, squamous cell carcinoma and adenocarcinoma, as well as stomach and colon cancer.
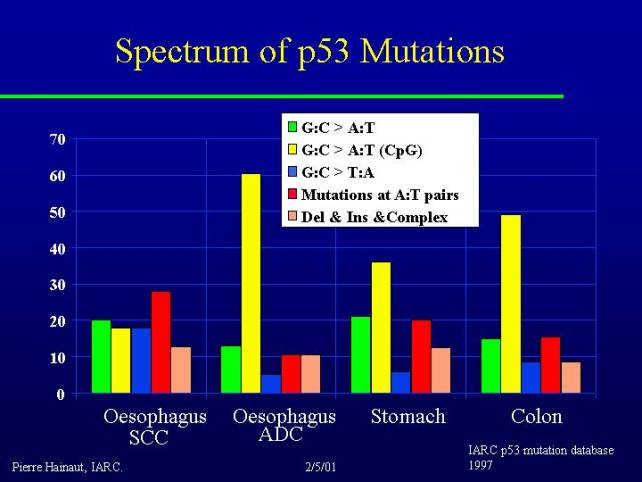
What you can see is that you find very different profiles: adenocarcinoma of the esophagus and cancer of the colon are dominated by CpG mutations (endogenous spontaneous mutation types). Indeed these two types of cancer have some similarities because they both can arise from preneoplastic lesions. It is interesting to note that there is a similarity between the profile of mutations between these two tumors.
If you look at squamous cell carcinoma in various parts of the world you find very big differences:
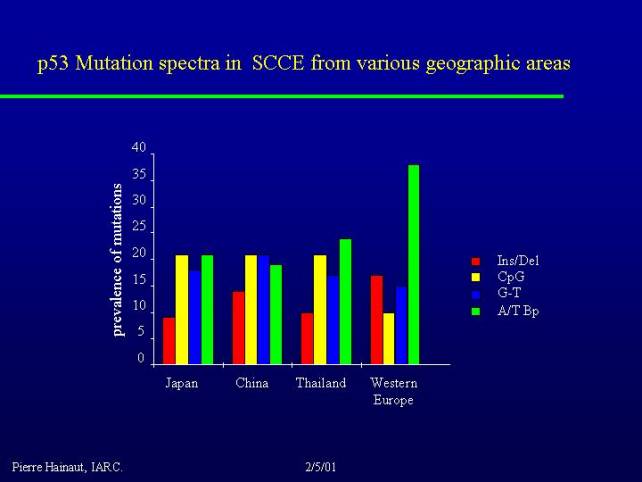
For example if you take high risk regions for this cancer such as Central China, you find very different mutations that you find in Western Europe. The mutations that occur at A to T basepairs for example, we can ascribe them to a direct effect of some metabolites of alcohol such as acetaldehyde. These mutations are much less common in the regions where alcohol is not thought to play a major role. So even if it is the same pathology you can find different types of mutation, which may reflect different risk factors.
This epidemiology of p53 mutations allows us to draw some conclusions about what might be the cause of these cancer:
I will show you two pieces of data about tobacco:
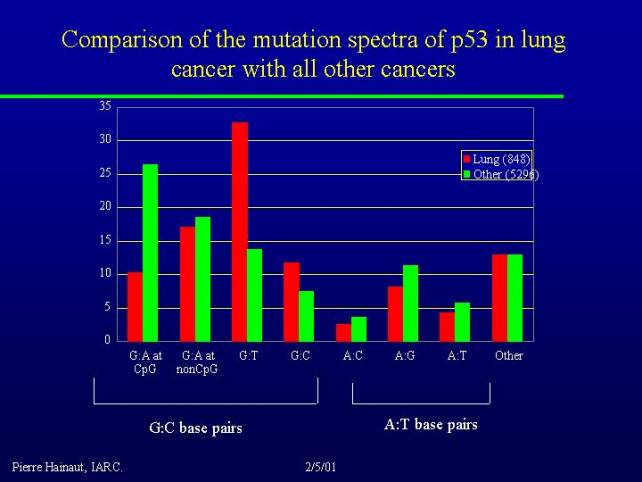
This is a comparison of the spectrum of mutations in lung cancer versus all other cancers. Many of G to T transversions are due to the direct mutagenic effect of the metabolite of benzopyrene, and you can see that they are much more frequent in lung cancer than in other cancers.
Even in lung cancers if you compare the data that we have for smokers and non-smokers, you can see that these G to T transversions are present in smokers but they are relatively rare in nonsmokers. So there is a clear association between that type of mutation and exposure to tobacco.
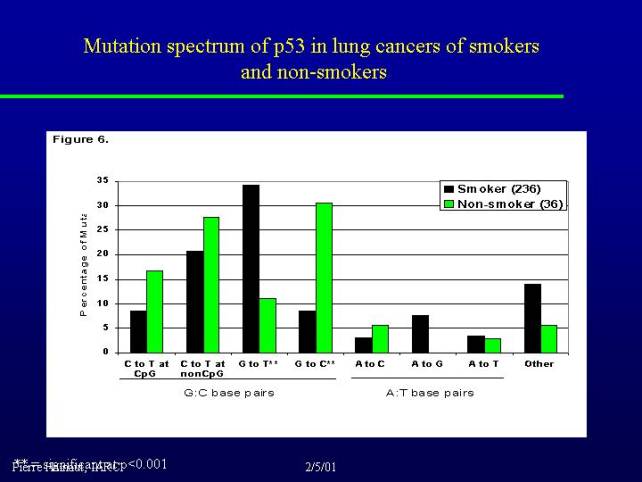
It is interesting to note that in nonsmokers we have another type of transversion (G to C). This remains to be evaluated on a larger number of individuals but these kind of mutation might be due to some other environmental exposure, as for example air pollution in cities, and this is currently a point that we are trying to evaluate.