Genetics
of retinoblastoma
by Dietmar
Lohmann
This presentation will review the genetics of retinoblastoma (RB) and the clinical aspects of this disorder, especially those with relevance for predictive testing in children at risk. The main part of this presentation will provide a detailed view of molecular genetics of RB, and the last part will introduce several practical points important for genetic counseling.
Clinical aspects
1. Presentation
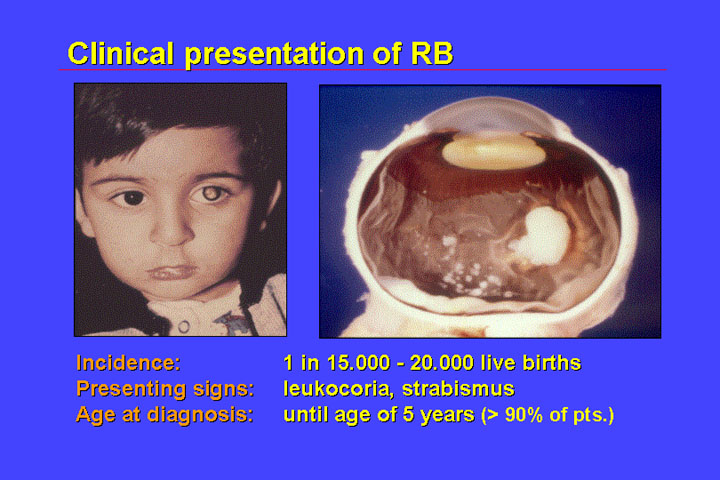
Retinoblastoma is a malignant tumor of the eye. It is rare, with an incidence of only 1:15-20.000 live-born children; affected children will develop retinoblastoma very early in life because retinoblastoma originates from cells that differentiate and are therefore not present in the adult organism. Therefore, retinoblastoma in the adult is not observed (except when it arises from a related, benign lesion termed retinoma).
The presenting signs of retinoblastoma are:
Age of diagnosis is early in life. Most cases are diagnosed before 5 years of age and only rarely is RB diagnosed in a child older than 5 years.
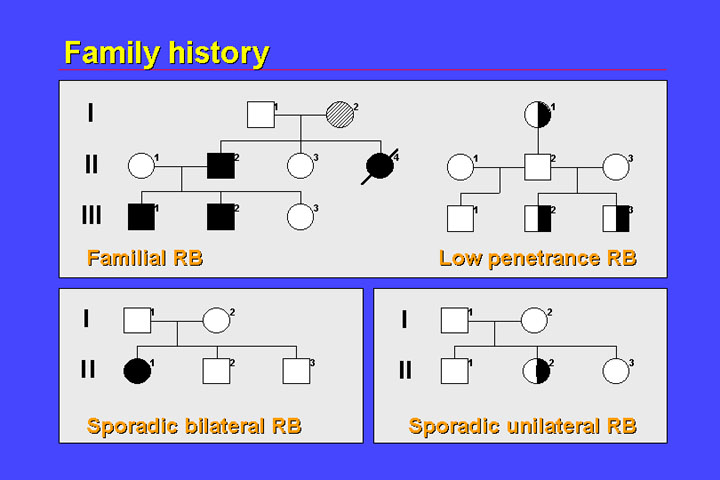
2. Family history
A second important
aspect of retinoblastoma is family history. In 10% of children diagnosed with
retinoblastoma,other relatives are also affected by this rare tumor.
This raises the important point that for any child with retinoblastoma, the relatives have to be screened for the presence of the benign tumors (retinoma), because presence of these benign tumors can indicate a familial form of retinoblastoma.
In most familial cases of retinoblastoma, the penetrance of retinoblastoma is nearly 100%. There are however some rare exceptions, the so called low-penetrance retinoblastoma families.
Families with unaffected carriers (that is reduced penetrance), also most often show reduced expressivity. Instead of bilateral disease - which is usually found in familial retinoblastoma - some patients will have only unilateral retinoblastoma.
Approximately 10% of children with retinoblastoma have familial disease. In 60% there is no positive family history, 30% of children have sporadic bilateral retinoblastoma. Most children have sporadic unilateral retinoblastoma.
3. Management of patients
The management of patients is important because retinoblastoma is a malignant tumor, and if therapy is delayed the tumor will metastasize with potentially fatal consequences.
Local therapy is adequate for treatment of retinoblastoma if the tumors are small. The tumor can be destroyed by freezing with cold sticks. Additionally, laser coagulation or radiation therapy (radioactive plug on the exterior of the eye, or by external beam radiation) can be used. As a last resort, the eye can be enucleated, which is very effective in preventing tumor spread.
After primary therapy of the tumor foci that have been observed at time of diagnosis, it is important to further observe the child:
The Molecular genetics of Retinoblastoma
1. Two hit hypothesis
Retinoblastoma was a model for Knudson's two hit hypothesis. In his initial hypothesis he stated that two mutational events are required for the initiation of retinoblastoma. Later it was shown that these two mutational events inactivate both alleles of a single gene (the retinoblastoma (RB) gene).
Hereditary retinoblastoma comprises all familial cases, almost all of the bilateral cases and some of the unilateral cases. In hereditary retinoblastoma the first mutation has been either transmitted from an affected or unaffected mutation-carrying parent - or has occurred de novo in the germline. Most often the paternal germline carries the de novo mutation.
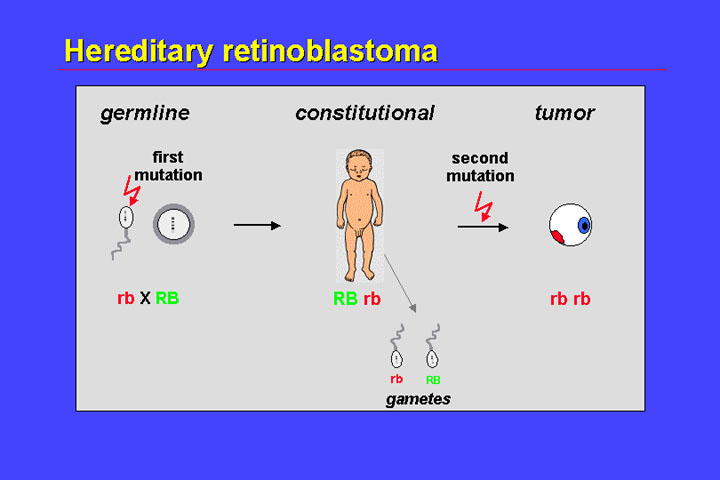
The child's cells - in all tissues - are heterozygous for this mutation. A single additional mutation is sufficient to inactivate both alleles, and give rise to the tumor. Because of independent second mutations in different retinoblastoma precursor cells, multiple tumor foci can occur. As a child is heterozygous, the germline of this child is also heterozygous with a corresponding 50% risk of transmitting a mutant-carrying gamete to its offspring. Therefore there is a recurrence risk of 50%.
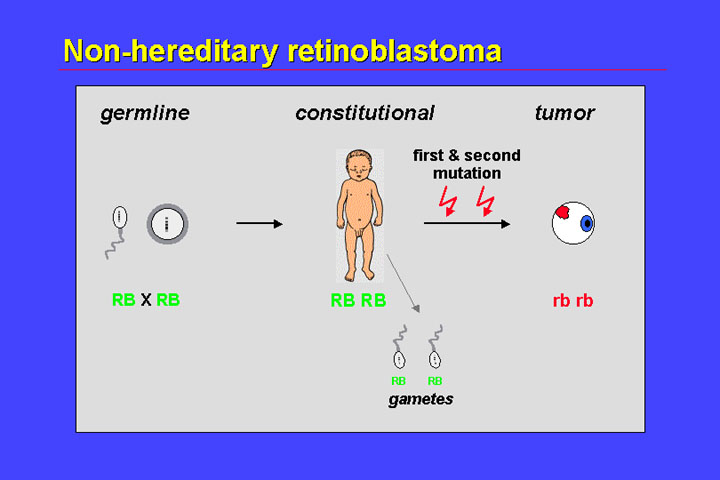
In non-hereditary retinoblastoma the first mutation is also a somatic event; two somatic mutations that inactivate both alleles of the RB1 gene in a single cell result in complete loss of the retinoblastoma gene in a precursor cell and thus initiate retinoblastoma development. The offspring of patients with nonhereditary retinoblastoma of course have no risk of retinoblastoma because the gametes do not carry a mutant allele.
An important phenomenon in retinoblastoma is loss of heterozygosity (LOH). LOH refers to the loss of one allele of a polymorphic locus owing to a second somatic mutation (generally, a relatively large deletion).
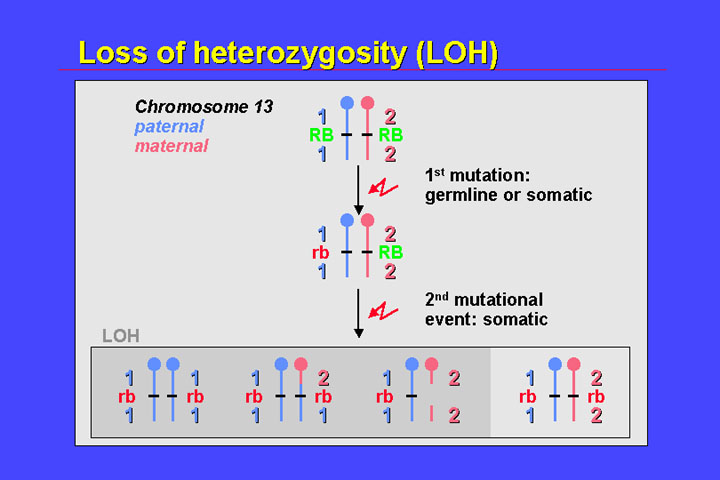
In the figure, the first mutation is located on the paternal chromosome (blue) and may be a point mutation that does not affect heterozygosity of nearby markers. The second mutation affects the maternal chromosome (pink) and leads to the loss of a significant segment of the long arm of chromosome 13. Consequently, only the paternal alleles of polymorphic markers in this region are visible.
There is loss of
heterozygosity for centromere-near and telomeric polymorphic loci. The most
frequent mechanism of loss of heterozygosity in retinoblastoma is mitotic recombination.
This is indicated by loss of heterozygosity at telomeric loci (heterozygosity
at the centromeric loci is retained).
2. The RB1 gene and its function
The retinoblastoma
gene was cloned some 14 years ago and is quite a large gene: the exons of the
retinoblastoma gene cover some 180 kB on chromosome 13q1.4.
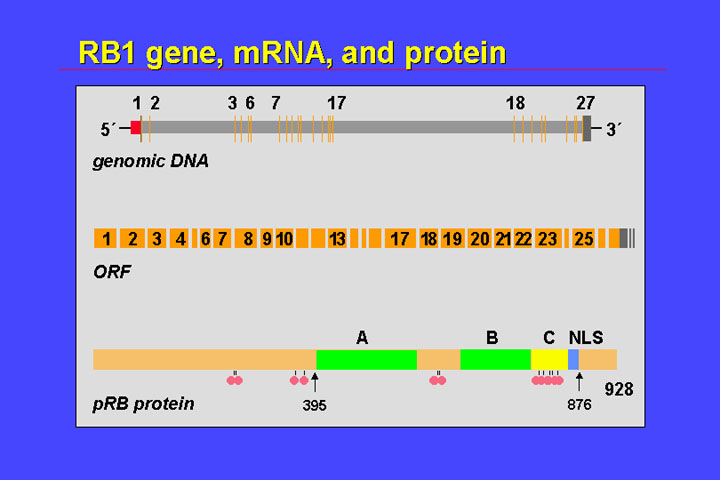
At the 5' end there is a CpG island, that normally is not methylated. It has 27 exons, the largest of which is smaller than 200 bp. The 27 exons have a coding region of 2.7 kB
They code for a nuclear phosphoprotein. It comprises a nuclear localization signal, and a potential phosphorylation site. Most important for the function of the retinoblastoma protein are the A and B pocket-domains. With these domains the retinoblastoma protein binds to the E2F transcription factor and binding of hypophosphorylated pRB to these factors represses transcription of the genes. If pRB is phosphorylated, it loses its affinity for E2F, E2F is released - and in consequence responsive genes are expressed. This switch between hypophosphorylated pRB and phosphorylated pRB is one part of the control of the restriction point which controls entering or transit from G1 to the S phase of the cell cycle.
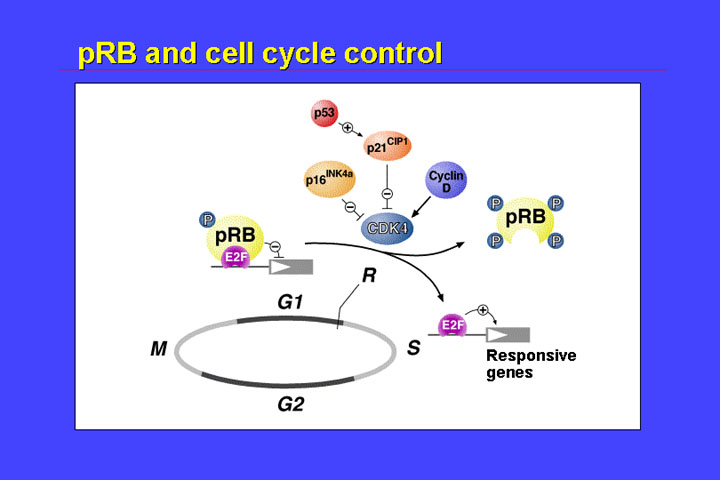
If pRB is lost due to mutations then affected cells are permanently lack restriction point control connected with pRB function. Therefore, it is to be expected that oncogenic RB gene mutations result in loss of function of the retinoblastoma protein.
3. Spectrum of oncogenic mutations

This in fact is the case. The spectrum of RB mutations that are observed in patients with RB and in tumors is composed largely of these different classes:
There is something special about patients with cytogenetic deletions because these patients can show - if the deletions are quite extensive - a 13 q deletion syndrome.
This is seen in about 5% of bilateral and 3% of patients with unilateral retinoblastoma, and importantly it is not uncommon to find that the mutation - the cytogenetic deletion - is present as a mosaic, especially in unilateral patients.
In addition to retinoblastoma these patients may show developmental delay, mental retardation and facial dysmorphism. Facial dysmorphism is not specific for the 13q- syndrome, and different patients with 13q- share only a few common features.
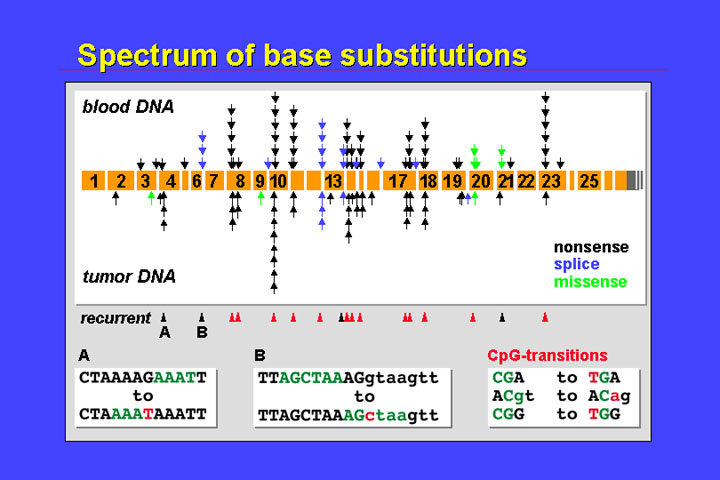
The figure represents the different locations and functional consequences of mutations that have been identified in peripheral blood DNA from patients with bilateral retinoblastoma, and in tumor DNA.
The most important class of mutations are single base substitutions. Roughly speaking the spectrum of mutations that might have occurred in the germline and of somatic mutations are quite identical.
There is independent occurrence of mutations at certain spots:
There is a predominance of nonsense mutations (black arrowheads), and missense mutations (green) are relatively rare.
The predominance of alterations that result in premature termination codons is also seen in the spectrum of small length mutations:
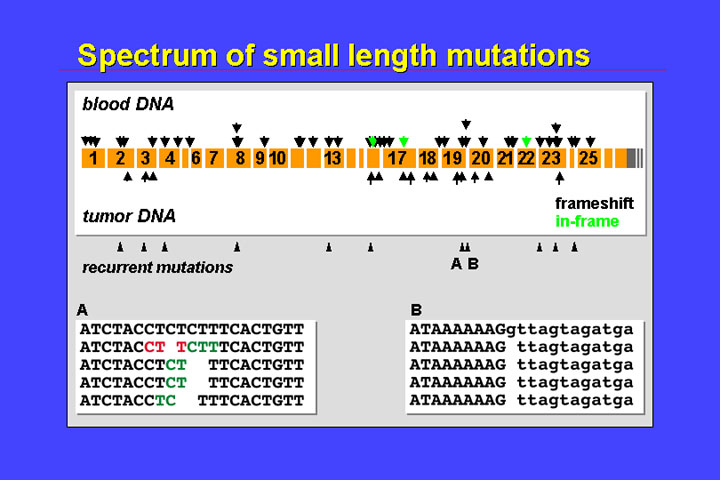
There is also some
recurrence and, as in the base substitutions, mutations are scattered all over
the gene. Single base substitutions and small length mutations have been observed
in all but the last two exons. Also reflecting the importance of endogenous
mutational mechanisms, recurrent mutations are observed at those sites that
are prone to polymerase slippage during replication (e.g., polypurine stretches
or repetitive structures such as CTCTCT…).
4. Genotype-phenotype associations
As far as genotype-phenotype correlation is concerned it is important to distinguish between the mutations that result in frameshift or nonsense mutations (i.e., mutations that result in premature stop codons), and in-frame and missense mutations (i.e., mutations that result only in local alterations).
The phenotypes associated with null alleles - that is alleles that have a premature termination codon - is very homogenous.
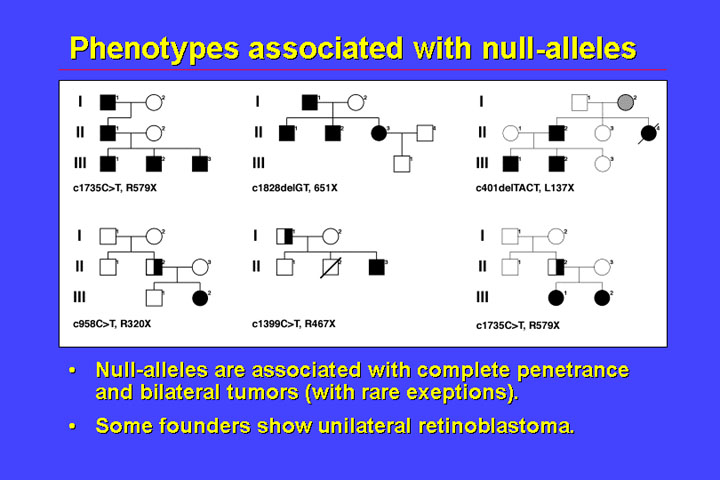
In families it
is normal to find complete penetrance for bilateral retinoblastoma. There is
an exception to the rule that patients who have mutations which cause a premature
stop codon have bilateral retinoblastoma - and that is the founders (or the
first affected) in a family. A part of them have only unilateral retinoblastoma
- and the reason for this is shown by the next figure:
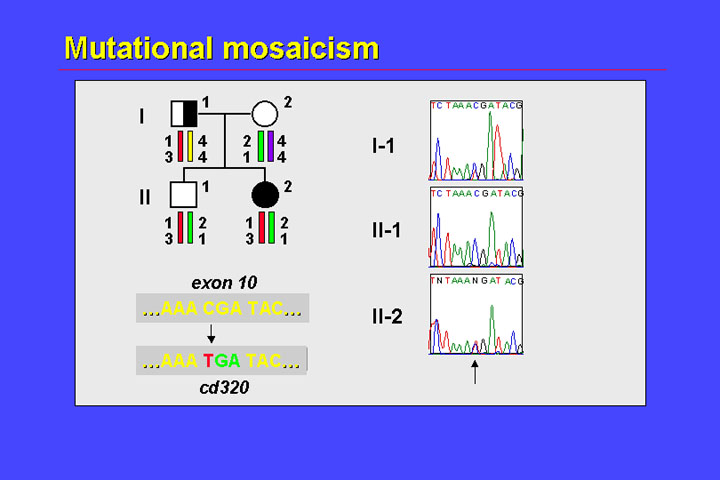
In this family, the father has unilateral retinoblastoma, the parents of this father were unaffected, his daughter has bilateral retinoblastoma, and his son is unaffected. Several lines of evidence suggest that the father displays germline mosaicism for this mutation:
This constellation may be regarded as strong evidence for germline mosaicism (i.e., the presence of two clones in germline cells, one of which is heterozygous for the mutation and one of which is homozygous wildtype).
Germline mosaicism
in founders appears to be a relatively common phenomenon, so that we have to
extend the timing of the first mutation graph by incorporating a mutational
event that occurs during embryogenesis.
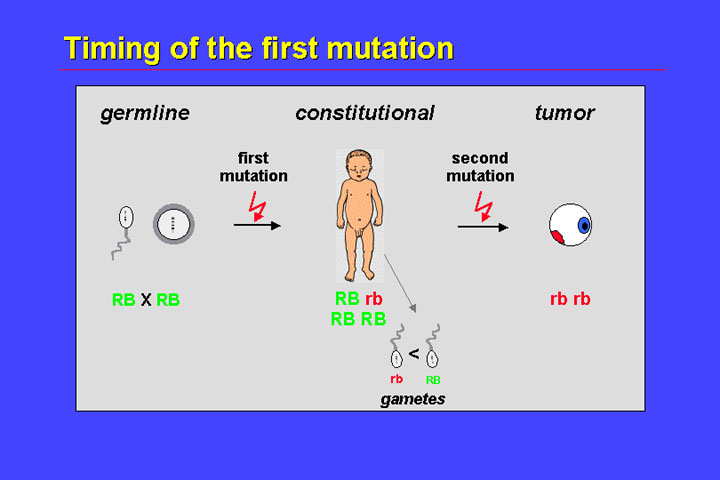
So not a germline, but an embryonal mutation results in constitutional mosaicism (one cell clone being heterozygous for a RB mutation and the remaining cells being wildtype). A second mutation in the mutant clone will result in tumor formation. Only if the germline of this mosaic carrier is affected, will there be transmission of mutant alleles to the offspring. To summarize this, unilateral retinoblastoma in founders is an example of reduced expressivity.
Additionally, there
are families with low penetrance:
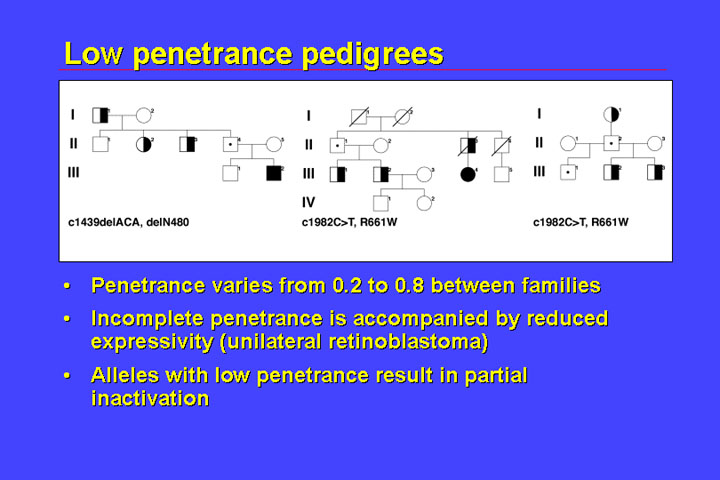
Low penetrance families generally have special kinds of mutations, that do not result in premature termination codons, but rather mutations that result only in local alterations. There are many other examples showing that low penetrance is associated with a distinct spectrum of mutations. In addition to reduced or incomplete penetrance there is reduced expressivity, that is there are not only unaffected carriers but those who are affected are most often affected by unilateral retinoblastoma. There is a clustering of mutations in the domains that code for the pocket A and B. This class of low-penetrance mutations results in structural changes of the protein.
There is a second class of low penetrance mutations:
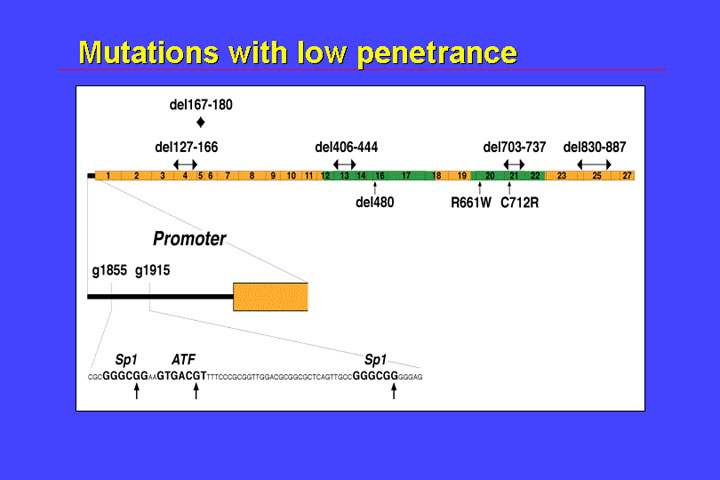
These mutations affect the promoter region, or the binding sites of SP1 and ATF (a transcription factor related or identical to CREB1.
In summary, expressivity and penetrance are linked together. Expressivity and penetrance of retinoblastoma depend on the functional consequence of the predisposing mutation:
Mosaicism is another cause of reduced expressivity depending on the number of cells and types of cells that carry the mutant allele.
Genetic counseling and predictive testing
Molecular risk prediction can be very complicated, and all relatives at risk of retinoblastoma should receive counseling.
1. Formal risk prediction
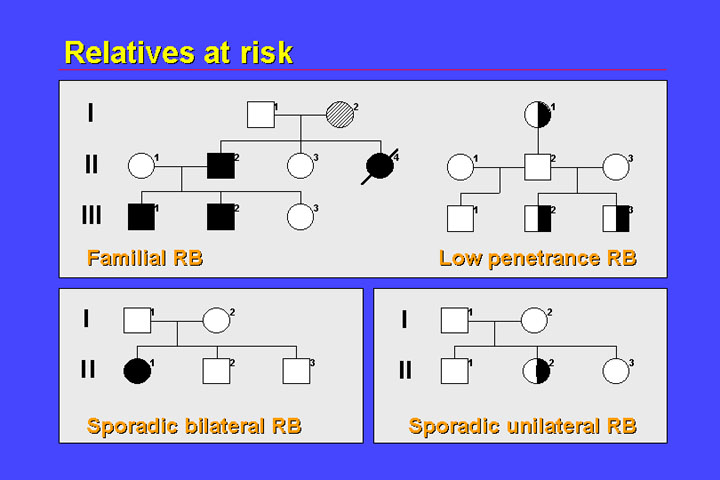
2. Molecular risk prediction
Molecular techniques allow more precise risk prediction. Without molecular analysis, children at risk have to be followed regularly. Practically this means that if a child is born to a parent with retinoblastoma, be it unilateral or bilateral, in the first few weeks of life - for example we usually have the second week of life - there is a first examination under general anesthesia. This is to be repeated in the first 3 months of life every 3 weeks. So every 3 weeks the parents have to come with their child to the ophthalmologic department, the child has to undergo general anesthesia and is searched for the presence of retinoblastoma. Therefore it is very important to exclude risk if it is possible.
a. Familial cases
In familial cases we can use indirect and direct testing. Both of these modalities are shown in the figure.
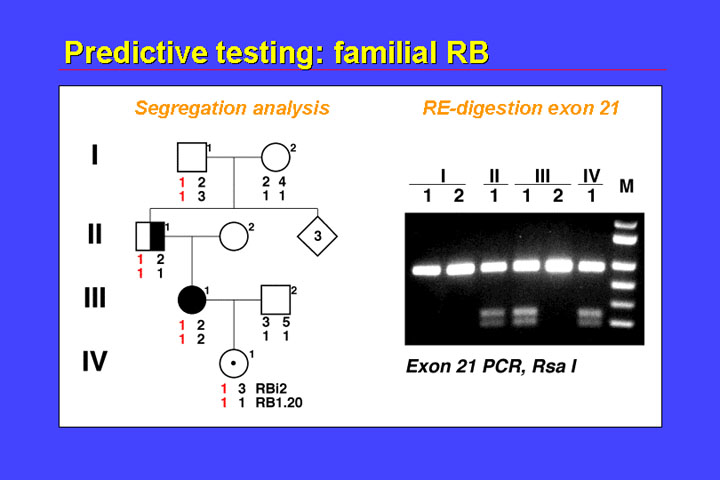
With indirect testing (typing of linked polymorphic loci), you can see that the 1.1. haplotype is in phase with the mutation. As this haplotype 1-1 has been passed from the mother (III.1) to the child (IV.1), it is to be expected that this child will develop retinoblastoma. We identified the mutation - in the mother, and this mutation can be identified by Rsa 1 digestion of exon 21 products, and you can see that II.1 is heterozygous, the mother is heterozygous, the father - of course - does not show the pattern, and the child is also heterozygous. We obtained this result about 8 hours after this child was born. So on the same day that this child was born we knew that it had to undergo surveillance because of the high risk of retinoblastoma.
b. Sporadic unilateral retinoblastoma
Genetic molecular risk prediction in unilateral retinoblastoma is most often dependent on the availability of fresh-frozen tumor sample.
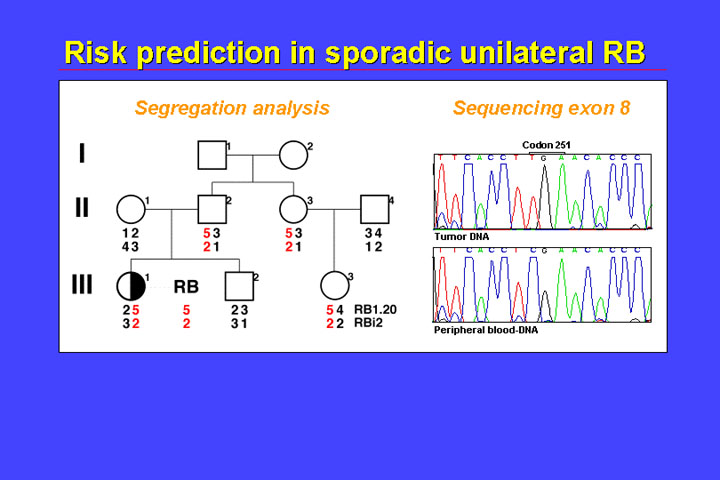
In the family shown in the figure, we first used indirect testing and there was loss of heterozygosity in the tumor (i.e., just the allele containing the mutation at codon 251 is visible). If the child III.1 has a constitutional mutation, then the mutation has to be in phase with the 5-2 allele. The 5-2 allele was inherited from the father, so we cannot exclude with indirect testing that the father has a mutation in phase with the 5-2 allele. The 5-2 allele is also present in the sister of the father and has been passed to this girl child, but not to the sibling (the sibling has the 3-1 haplotype). Therefore, indirect testing allows us to exclude the about 2% risk in the sibling (III.2), but cannot exclude the very small risk in the cousin (III.3).
Other links: