
Genetics
of Neuroblastoma
by J.
Maris
Clinical features
Neuroblastoma is an embrional cancer like retinoblastoma and nephroblastoma (Wilms tumor). The precursor cell is an undiferentiated neuroblast from the neural crest. It is a true pediatric disease with the median age of diagnosis less than 2 years. Essentially 95% of patients are diagnosed by age 5 and it is very rare to see teenagers with the disease - although it does occur. Neuroblastoma is still an important cause of childhood cancer mortality.
The clinical heterogeneity is a fascinating topic and I would like to begin this presentation with two exemplary cases:
In addition, neuroblastoma remains an important clinical problem for pediatric oncology. Whereas there has been enormous success over the last several decades in improving the outcome for many childhood cancers, there has only been moderate success in improving the outcome for children with neuroblastoma. In 1995-2000, only half the children who are diagnosed with the disease are expected to survive.
The clinical heterogeneity is a theme I would like to develop. We can broadly group children with neuroblastoma in 3 categories:
In addition to the clinical heterogeneity, neuroblastoma displays a high degree of genetic heterogeneity:
|
|
We still do not know what events initiate tumorigenesis. There is a broad range of somatically aquired alterations that can broadly be grouped into: loss of genetic material as well as gain of genetic material at the n-myc locus in the long arm of chromosome 17 (see text box).
Constitutional rearrangements
Constitutional rearrangements have been very important in the cloning of predisposition genes for both retinoblastoma and Wilms tumor. In neuroblastoma constitutional abnormalities have only been very rarely identified.
Associated conditions
In addition there are very few associated congenital abnormalities in patients with neuroblastoma. There is a list of syndromes that are seen with Wilms tumor, and those went a long way towards helping to identify the WT1 gene - in fact were the reason why WT1 was cloned. There are rare patients with not only neuroblastoma but other disorders of neural crest derived tissue.
These include:
This has suggested that perhaps at least some of neuroblastomas can be explained by a more global disorder of neural crest derived tissue. However, there is no single associated condition that would be usefull in identifying neuroblastoma predisposition genes.
Hereditary neuroblastoma
Hereditary neuroblastoma does occur, however it is excedingly rare, and a recent survey of French data suggested 1.4 % of newly diagnosed cases of neuroblastoma have a positive familial history for the disease.
In 1972 (one year after his seminal paper on hereditary retinoblastoma), Knudson reviewed data on hereditary neuroblastoma. He noted that - like retinoblastoma - familial neuroblastoma showed an autosomal dominant mode of disease transmission, the patients in these families were diagnosed in earlier median age, and they very often had multiple primary tumors - the classic hallmarks of hereditary predisposition cancer syndrome.:
However in distinction to retinoblastoma, neuroblastoma is incompletely penetrant. In large part it is now clear that this is due to the fact that neuroblastomas sometimes spontaneously remit and just never come to clinical attention. In adition, neuroblastoma - unlike retinoblastoma and Wilms tumor - may often be fatal. Therefore large pedigrees are not common.
This is a large pedigrees with a typical autosomal dominant mode of disease transmission. In this families, as well as several others, neuroblastoma cosegregates with Hirschsprung disease (for instance, IV.5)
Consistent with Knudson's prediction, most of these children were diagnosed very early in life, and several of them had multifocal tumors (more than one primary tumor), consistent with the two hit model. We and others have spent a lot of time looking for a neuroblastoma predisposition gene, and there were many candidates for a neuroblastoma predisposition gene.
|
Linkage analysis of hereditary NB 13 candidate regions excluded (lod < 2.0) - LOH: 1p36, 3p21-pter, 4p16, 11q13-24, 14q32 - Gain: MYCN, 17q23-qter - Neurocristopathy: RET, EDNRB, EDN3, NF1, GDNF, TRN-R2 Genome-wide scan for linkage: |
First it seemed obvious to me and others that this gene must be at 1p36 because there are constitutional deletions syndromes there - or patients with constitutional deletions there - but this region as well as many other regions were shown not to be consistent with hereditary neuroblastoma or linked to that phenotype. A recently completed genome wide scan suggests that the short arm of chromosome 16 contains a hereditary neuroblastoma predisposition gene. The following figure gives an example of a pedigree demonstrating linkage to this locus (The red number indicates that DNA was available for genotyping).
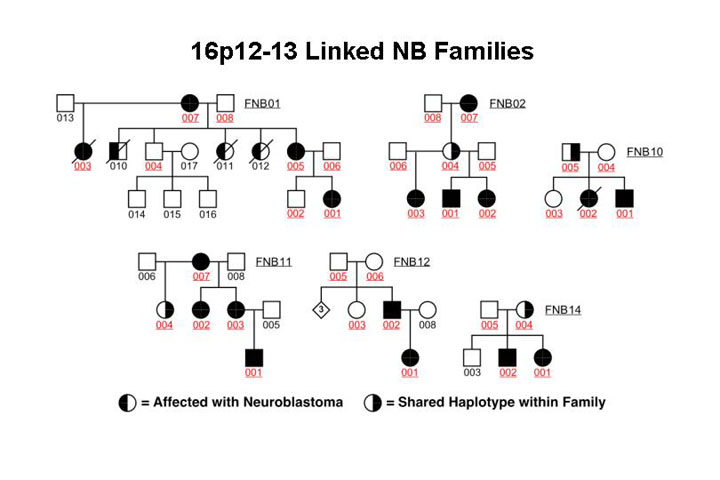
This sort of data
was highly suggestive of chromosome 16 being consistent with containing a predisposition
gene but it is important to note here that not all families show linkage to
16 p and it appears that there will be more than one hereditary neuroblastoma
predisposition gene.
If this predisposition gene is a tumor suppressor one would expect that the
non disease associated alelle would be deleted in at least a subset of the tumors.This
is shown in the following figure
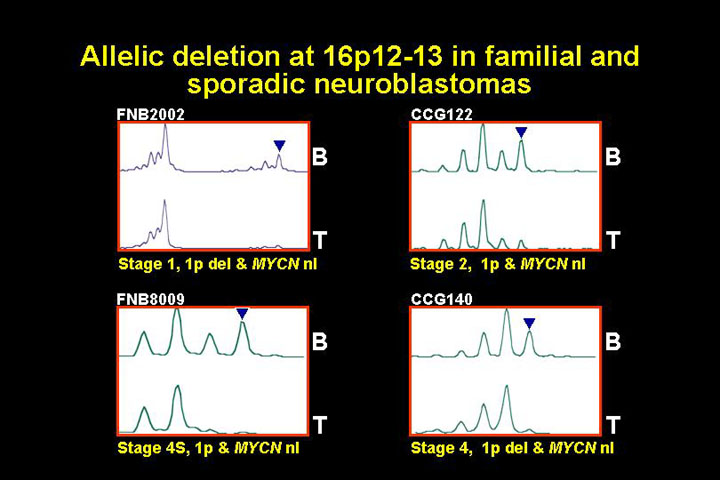
The following figure shows loss of heterozygosy data for two different tumors from patients with hereditary neuroblastoma.

This is just some representative data of loss of one of the alelles in the tumor, suggesting that this would be a classic tumor suppressor on chromosome 16.
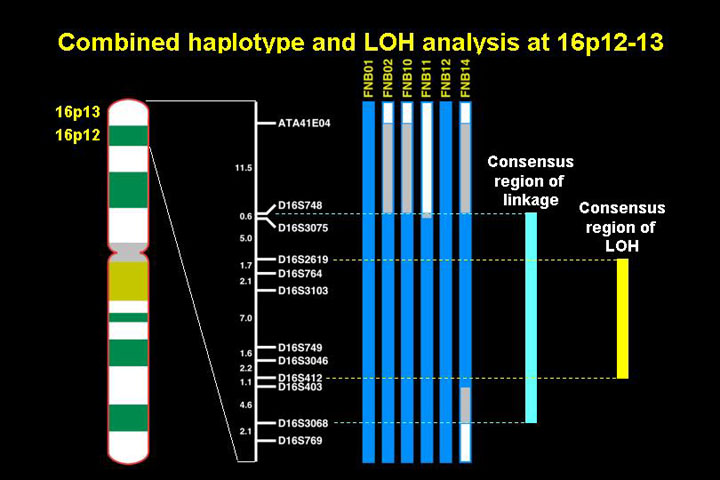
The current status (2000) of the mapping of the neuroblastoma predisposition gene on chromosome 16 is that a the common haplotype that is seggregating with the disease on chromosome 16 has been iodentifdied in some of the families on the distal short arm of chromosome 16. This overlaps with the region of LOH (12 cM) so that this region is likely to contain the hereditary neuroblastoma predisposition gene.
Somatically acquired chromosomal alterations
This is a list of regions of the genome that are recurrently lost or gained in tumor material from neuroblastoma patients.
|
Acquired Genomic Alterations in Primary Neuroblastomas Loss of genetic material - 1p36: 19-36% Gain of genetic material - MYCN amplification: 20-25% |
I. Loss of genetic material
1. Chromosome 1
The short arm of chromosome 1 has received a lot of interest by several laboratories. Deletion of this region is very common in neuroblastoma and classic suppression-of-tumorigenicity studies with microcell mediated transfer showed that replacement of chromosome 1 in cell lines that were missing chromosome 1 greatly suppressed tumorigenicity.
This observation together with the fact that there are constitutional deletions at chromosome 1p suggested that chromosome 1 contains a tumor suppressor gene important for neuroblastoma tumorigenesis. The cloning of this gene has been the focus of many labs, but the gene has not been cloned. Currently there is about a 1 megabase region where there are 23 or 24 transcripts, and no mutations in the retained allele have yet been identified.
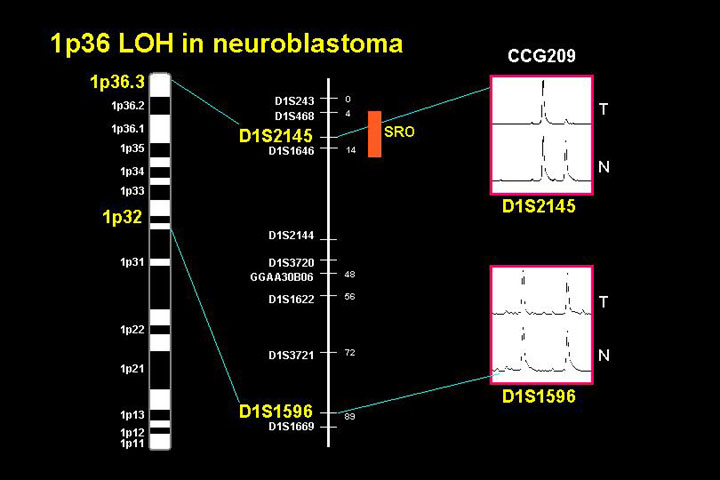
This figure shows a cytogenetic map in the region of consistent loss of heterozygosity and on the right the region of deletion defining the relatively narrow interval where a neuroblastoma suppressor gene is thought to be located. It is not clear why this gene has not been cloned yet, and one supposition is that perhaps this is not a classic tumor suppressor where the alternate allele would have mutations.
One of the reasons for a lot of interest in chromosome 1 is that deletion of this region is associated with patients who have high risk disease. In general, about one third of neuroblastomas display 1p deletions.
Table: 1p36 Clinical Significance (1p36 LOH: 83/238 [35%])
| Variable | LOH 1p36 | P value |
| Age > 1 year | 67/171 (39%) | 0.026 |
| INSS Stage 4 | 52/109 (48%) | <0.001 |
| Shimada UH | 56/117 (48%) | <0.001 |
| Ferritin > 142 | 35/70 (50%) | <0.001 |
| MYCN Amp | 44/57 (77%) | <0.001 |
There is a very strong correlation with many of the features suggestive of a high risk phenotype: age greater than 1, metastatic disease at diagnosis and some other features such as N-MYC amplification that predict an aggressive tumor course. Additionally, event-free survival (EFS), relapse status and overall survival (OS) all are associated with 1p status (see figure).
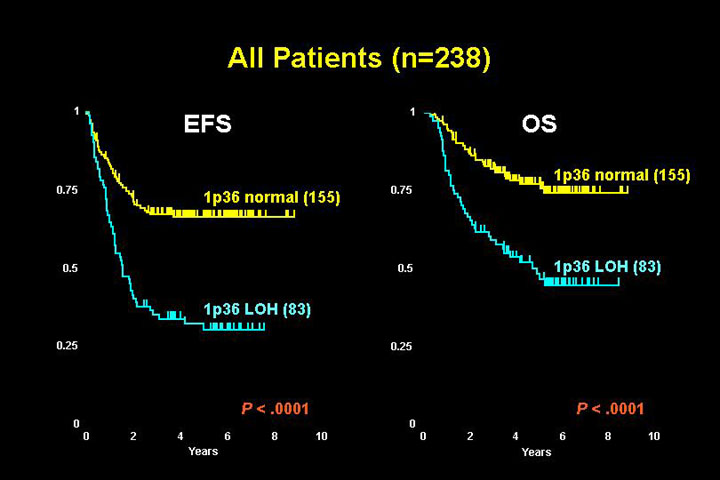

Multivariate analysis clearly shows that 1p36 loss of heterozygosity is independently prognostic for disease relapse, so it is an important marker of high risk disease.
2. Chromosome 11
Many patients with neuroblastoma have deletions of chromosome 1 but other regions are deleted frequently. Unlike chromosome 1 where almost all the deletions appear to be restricted to the region from 1p32 to the telomere, chromosome 11 deletions fall into two broad categories.
The following figure shows LOH data from both the short arm of chromosome 11 as well as the long arm of chromosome 11.

About half of patients have a normal chromosome 11 status in their tumors, but another half have deletions of chromosome 11 that fall into two patterns:
On the long arm, if you map many different tumors with deletions, a common region of deletion has been identified in 11q23.3

This region is 2 or 3 Mb in size, and is currently the focus of positional cloning efforts.
11qLOH: Clinical and Biological Correlations
| Variable | P value 11q LOH |
P value
Unb[11q] LOH |
| Age 1-5 years | 0.813 | 0.023 |
| Stage 4 | 0.214 | <0.001 |
| Shimada UH | 0.706 | 0.002 |
| 1p36.3 LOH | 0.090 | 0.490 |
| Unb 17q gain | 0.004 | <0.001 |
| MYCN Amo | <0.001 (inverse correlation) | 0.007 (inverse correlation) |
There appear to be subsets depending on how 11q23 deletions originate in neuroblastoma. The mere existence of an 11q23 deletion (which is found in about half of all patients) does not correlate with any of the standard clinical features. However, there is a very strong inverse correllation with N-MYC amplification.
Neuroblastoma associated with an unbalanced rearrangement resulting in loss of 11q23 defines a group of patients that are at much higher risk, but is also inversely correlated to n-myc amplification.

3. Other regions of deletions
In the above, I have shown you examples of two regions of deletion where putative tumor suppressor genes are located. There is a lot of interest by many groups in other regions of deletions such as 14q as well as other regions where there appears to be a slightly less frequency but definitely recurrent abnormalities at these loci.
|
Allelic deletion at other loci and alterations in known tumor suppressor genes (TSGs) 14q32 LOH in 20-25% of cases: Associated with 11q23 LOH and inversely associated with MYCN amplification 3p14-24. 4p. 9p. 18q LOH in 15-20% of cases No de novo mutations found in TP53, CDKN2 or other known TSGs TP53 mutated in some relapse specimens in association with acquired drug resistance |
Alterations in known tumor suppressor genes (TSG)
P53 - the most commonly mutated gene in human cancer - is never mutated in neuroblastomas at diagnosis. P16, or other known tumor suppressor genes are only very, very rarely homozygously deleted or mutated. Some recent data show that in relapse specimens p53 mutations are actually very common actually. In neuroblastoma p53 may be a target and play an important role in aquired drug resistance. But de novo mutations are exceedingly rare.
II. Gain of genetic material
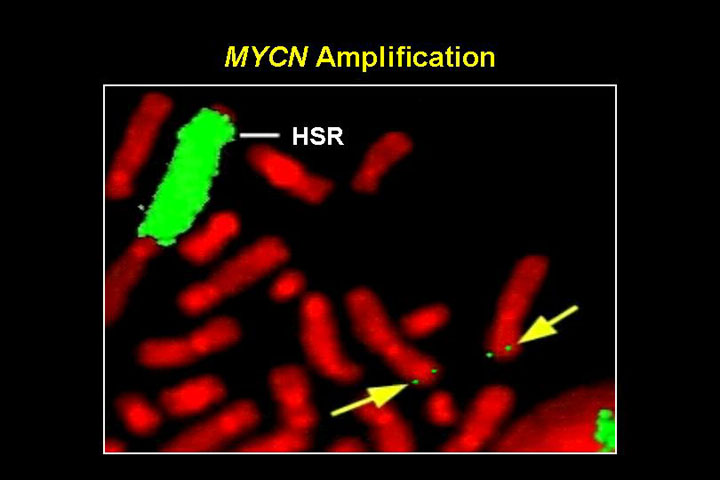
The normal n-myc locus on 2p24, and multiple copies of n-myc rearranged in a homogenously staining region

This figure shows a FISH experiment: with multiple copies of n-myc rearranged in a homogeneously staining region (HSR) on a different chromosome - one of the classic ways in which n-myc amplicons are formed. When n-myc is amplified, it is not subtle. There are 50 - 100 or 150 copies. This is present as double minutes within these cells and it is heterogeneous, some cells have lower level amplification, some very high amplification.
|
MYCN amplification in neuroblastoma 20-25% of
primary tumors |
Amplification of n-myc occurs in about 1/4 of primary tumors, it is highly associated with deletions of 1p. Nearly every tumor that has amplification of n-myc will have deletion of chromosome 1p. This amplification results in an elevated message and very high protein expression, and independently predicts for treatment failure. It is the only oncogene that is consistently overexpressed in neuroblastomas. Recent work with targeted overexpression of n-myc in the neuroectoderm in transgenic mice showed that these mice developed tumors similar to neuroblastoma, strongly suggest that n-myc plays a very important causal role in the development of neuroblastoma.
N-MYC has an important part in the history of molecular biology. In clinical oncology this was the first example of using molecular genetic data prospectively to stratify patients to therapy, and it is something that is still done today, it is one of the first things that is done with a neuroblastoma biopsy specimen - to see if it is N-MYC amplified or not.
A survey of 3000 neuroblastomas shows that the amplification is much more frequent in patients with advanced stage disease, and that correlates dramatically with survival.
MYCN amplification and survival in 3000 neuroblastomas
| Stage at diagnosis | Amplification | Survival |
| Ganglioneuroma | 0% | 100% |
| Low stage (1-2) | 4% | 90% |
| Stage 4S | 8% | 80% |
| Advanced stage (3,4) | 32% | 30% |
| Total | 22% | 50% |
This table is taken from the original paper by Seeger et al. who showed in 1985 that N-MYC amplification is highly associated with a poor disease outcome in many different patient subsets. These results are still valid today; for instance, infants with neuroblastoma and N-MYC amplification have a very poor survival probability whereas the vast majority of infants with single copy N-MYC are disease survivors. N-MYC has an important part in the history of molecular biology. In clinical oncology this was the first example of using molecular genetic data prospectively to stratify patients to therapy.
The gene

N-MYC on the short arm of chromosome 2.
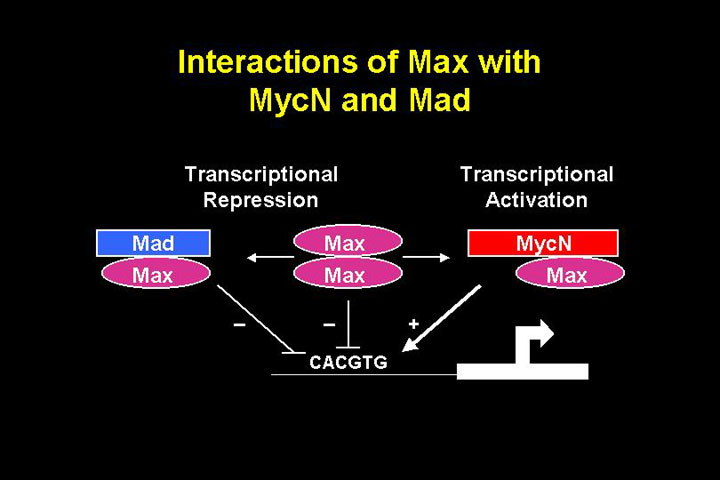
The protein


Max homodimers are transcriptionally repressive, Max-MYC heterodimers activate transcription. Therefore, if N-MYC is overexpressed it leads to transcriptional activation.
In the mid 1990's when comparate genomic hybridisation (CGH) came into broad use it was used to confirm the fact that N-MYC was amplified very frequently and chromosome 1 deletions occurred frequently. CGH also verified that there are many different chromosomal gains and losses that occur in human neuroblastoma.
One thing that really seemed to stand out was that gain of chromosome 17 was very frequent, which was more frequently noted in CGH experiments than was originally noted from kariotypes or other types of surveys.
|
Unbalanced Gain of distal 17q in neuroblastoma
|
Gain of the distal long arm of chromosome 17 was originally identified in cariotypes and noted to be recurrent, but the overall frequency was not apreciated. CGH experiments suggest that 50 - 75% of human neuroblastomas contain gain of chromosome 17, making it the most common genetic alteration in primary neuroblastoma. There is a 25-Mb common region of gain located at 17q23.1-17qter that is associated with adverse prognostic features.
Other abnormalities that occur in human neuroblastomas
This survey has been biased towards abnormalities that can be detected at the kariotype level by gains or losses. In the following, I will offer a short introduction to some of the other genetic abnormalities that occur in human neuroblastomas.
1) The neurotrophin signaling pathway
Neurotrophins
| Receptor | Ligand | Tumor biology association |
| TrkA | NGF | Favorable |
| TrkB | BDNF | Unfavorable |
| TrkC | NT3 | favorable |
2) caspase 8
Caspase 8 is a serin protease that is very important in the effector arm of extracelullar apoptotic signaling. It is homozygously deleted in a small subset of neuroblastomas, but more importantly - is functionally inactivated by hypermethylation in almost every neuroblastoma with n-myc amplification.
Like n-myc, caspase 8 can activate transcription. It also - like n-myc or c-myc - is important when overexpressed in pushing the cell towards apoptosis so it has dual functions, and this seems to obviate the apoptotic signaling that can occur when n-myc is overexpressed.
|
Alterations
in gene expression during
|

Model of the genetic basis of neuroblastoma
The following figure
offers a summary of the signaling pathways that may be involved in low risk,
intermediate risk and high risk neuroblastoma
On the left is a normal diploid neuroblast (2N). Certain genetic alterations
(3N or 17q+) or an inherited mutation in the neuroblastoma gene or in an as
yet unidentified gene on 1p gene act as a trigger for the initiation of the
development of neuroblastoma.
There seem to be two main pathways. The timing of the initial mutational event may determine which pathway is taken.
The final table offers a summary of the relevance of each of these parameters for biological staging of neuroblastoma.
Biological Staging
| Parameter | Type 1 | Type 2 | Type 3 |
| MYCN | Normal | Normal | Amplified |
| 17q23-25 | Normal | ±Gain | ±Gain |
| DANN index | Triploid | ±Diploid | Diploid |
| 1p36 | Normal | ±Deleted | Deleted |
| 11q23/14q32 | Normal | Deleted | Normal |
| Trk expression | Trk A | Trk A or B | Trk B |
| Survival | 95-100% | ? | <30% |