Introduction
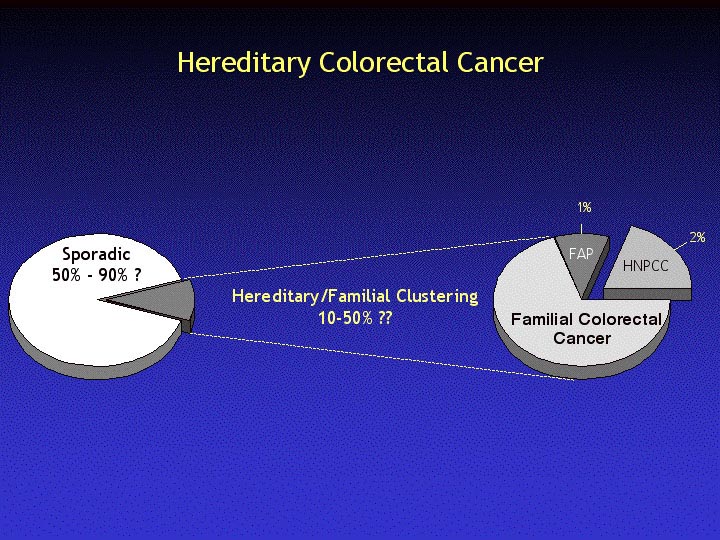
Colorectal cancer is still one of the major causes of morbidity and mortality in western, industrialized countries (Western Europe and North America). As it's true for most of the cancer syndromes, a percentage of the colorectal cancer cases have a strong familial basis.
There are two well defined syndromes, familial adenomatous polyposis (FAP; about 1% of the total cases) and hereditary nonpolyposis colorectal cancer (HNPCC; about 2% of the total cases). Yet, in the majority of the familial cases, the underlying genes are still unknown.
I. Familial Adenomatous Polyposis
Familial adenomatous polyposis, or FAP, is an autosomal dominant predisposition to the development of multiple adenomatous polyps in the colon and the rectum. Notably, FAP patients are at risk for a very broad spectrum of extracolonic manifestations (this is a feature of many inherited cancer syndromes).
The onset of the colorectal polyps is usually early (2nd - 3rd decade of life), and the penetrance is nearly 100%. However, vast clinical variability has been observed as far as the age of onset, number of polyps, as well as type and number of extracolonic manifestations are concerned.
Colorectal Polyps
The hallmark of FAP is represented by the occurrence of numerous colorectal polyps. The colonic mucosa is typically covered with hundreds to thousands of small polyps. The polyps in FAP are benign tumors with a very high risk of developing into malignant carcinomas upon invasion of the underlying muscularis mucosae.
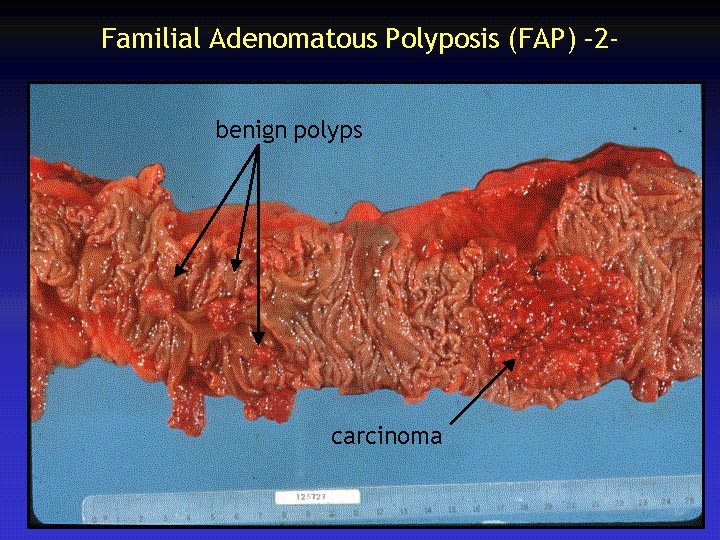
In the figure, several as yet benign polyps can be observed. However, one polyp has degenerated into a carcinoma, a potentially deadly lesion.
The APC gene [OMIM]:
FAP is caused by germline mutations in the APC gene. Since the manifestations of FAP can be of endodermal, mesodermal and ectodermal origin, it is likely that APC plays an important role in development, differentiation, and tumorigenesis.
The APC gene spans approx. 150 kb of genomic DNA and is fragmented into 15 exons which are spliced into an mRNA transcript of about 8 kb. The exon structure of APC is remarkable in that the first 14 exons are relatively small while exon 15 codes for an open reading frame of about 6.5 kb.
APC protein
APC encodes a large, 312 kD protein, which is both cytoplasmatic and nuclear. The amino acid sequence of APC is indicative of an extremely multifunctional protein.
The amino terminal domain of APC encompasses a coiled-coil domain is responsible for homodimerization. In the central domain, APC interacts with a very important cell adhesion and signaling molecule, b-catenin, through a series of binding- and downregulaing repeats. In its C-terminus, APC interacts with EB1 (a mitotic regulator in yeast), DLG (a Drosophila tumor suppressor gene) and with the microtubule cytoskeleton. The EB1- and microtubulin-binding domains were recently showed to be involved in the attachment of the mitotic spindle to the chromosome via the kinetochore anc cause chriomosomal instability (CIN) when mutated.
Kaplan, et al. (2001), A role for the Adenomatous Polyposis Coli protein in chromosome segregation Nature Cell Biol 3:429-432 - Abstract
Fodde, et al. (2001), Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol 3:433 - 438)- Abstract
APC - in Cancer
APC is now thought to be the gene for colorectal cancer: not only are germline APC mutations responsible for Familial Adenomatous Polyposis (GRODEN et al. 1991; NISHISHO et al., 1991), but somatic APC mutations are found in the vast majority (85%) of sporadic colorectal tumors regardless of histological stage (MIYOSHI et al., 1992; POWELL et al., 1992). This suggests that loss of APC function is likely to play a rate-limiting role in tumor initiation in the colorectum.
The capacity of APC to regulate intracellular b-catenin levels within the WNT signal transduction pathway represents its main tumor suppressing activity. In this signal transduction pathway, the WNT ligand binds the receptor, frizzled, and activates an inhibitory process thus allowing the accumulation of b-catenin in the cytoplasma and its translocation to the nucleus. Here, b-catenin associates with transcription factors of the Lef/Tcf family of HMG (high mobility group) family. In the absence of the ligand, a complex is formed between APC, b-Catenin, GSK3b (a Ser/Thr kinase), and the scaffold proteins Conductin and Axin. The formation of this complex leads to the phosphorylation of b-catenin. The latter is a signal for b-catenin ubiquitinization and proteolytic degradation. Therefore, if no WNT is present, β-catenin is degraded. In the presence of the WNT ligand, however, transcriptional activation of specific WNT target genes. Two different mutational mechanisms activate the WNT pathway in colorectal tumors:
Which WNT downstream target genes are responsible for colorecta cancer?
To date a limited number of downstream target genes have been identified: two of them, c-myc and cyclin D1, are implicated in biological processes such as cell-cycle regulation and apoptosis clearly related to with tumor initiation and progression.
Mutation analysis
In this section, mutation analysis of the APC gene will be discussed. Denaturing gradient gel electrophoresis (DGGE) and protein truncation test (PTT) are commonly used for the mutation analysis of this gene.
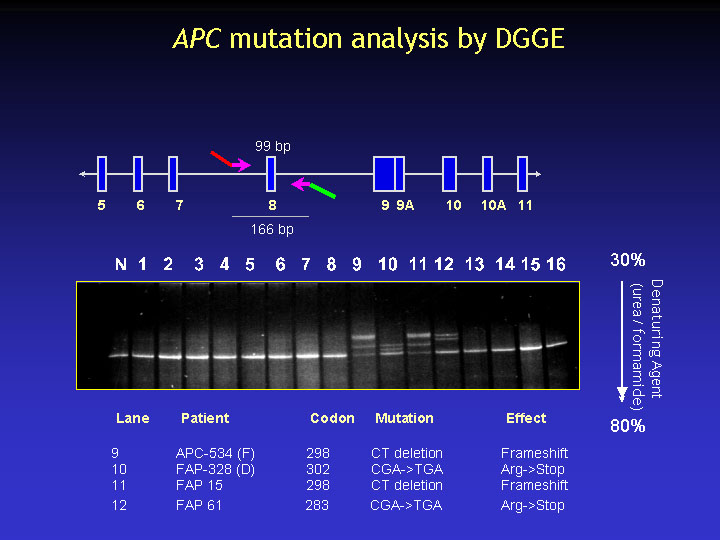
The figure shows an example of analysis of APC exon 8. The 4 bands pattern in the gel is indicative of the presence of a nucleotide variant (either a disease-causing mutation or other polymorphism). Sequence analysis of these samples usually reveals either frameshift or nonsense mutations that predict the truncation of the protein product.
Fodde, R and Losekoot, M. Mutation detection by denaturing gradient gel electrophoresis (DGGE). Hum Mutat 3 (1994) 83-94. - Abstract
Accurate mutation detection is very important in FAP, a disease that affects individuals in their 2nd or 3rd decade of life where pre-symptomatic detection of mutation carriers represents a life-saving tool. Once the carrier status has been established, colonoscopic surveillance at regular intervals is recommended to allow early polyps detection and prevention of mailignancies. Non-carriers within an affected kindred have the same CRC risk as the general population and can be spared the surveillance programme by colonoscopy.
The spectrum of the APC mutations is quite heterogenous. Mutations, mainly nonsense and frameshifts, are spread throughout the 5' half of the gene.
Protein truncation test (PTT)
The protein truncation test (PTT) is a simple test in which PCR- amplified cDNA or genomic DNA is translated into a protein in the presence of a labeled amino acid. The sample is then run on a SDS polyacrylamide gel, and the resulting protein products, either full-length or truncated, are detected by autoradiography. Truncated proteins are indicative of the presence of a mutation causing a premature stop codon.
van der Luijt R, Khan PM, Vasen H, van Leeuwen C, Tops C, Roest P, den Dunnen J, Fodde R (1994) Rapid detection of translation-terminating mutations at the adenomatous polyposis coli (APC) gene by direct protein truncation test. Genomics 20:1-4. - Abstract
Genotype - phenotype correlation
In view of the heterogeneity of both the APC mutation spectrum and of the corresponding clinical outcome, genotype-phenoty correlations have been successfully established at the APC gene in FAP.
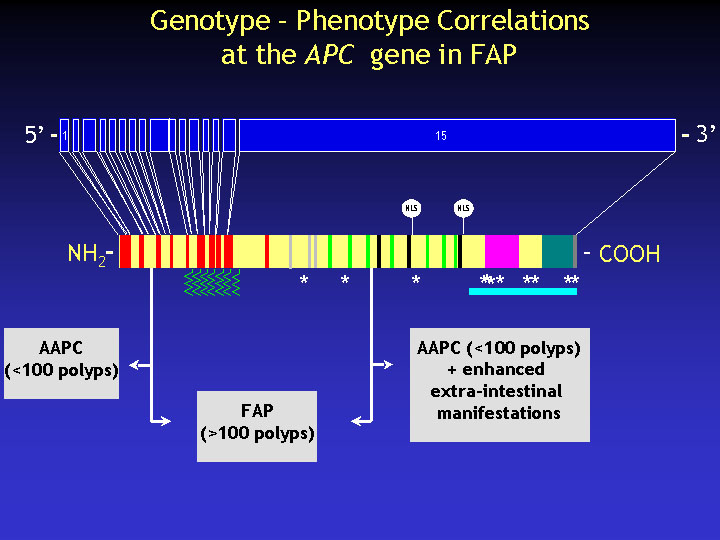
Mutations in the middle part of the gene are associated with 'classical FAP' i.e. the full-blown expression of the disease with early onset of hundreds to thousands polyps in the colorectum. Mutations in the extreme 5' and in the 3' half of the gene are associated with atypical or attenuated APC (AAPC), where patients develop less than 100 polyps, with a delayed age of onset. AAPC mutations in the 3' half of the gene are usually associated with enhanced extra-intestinal manifestations such desmoids and osteomas.Interestingly, whereas mutations leading to classical FAP usually lead to the stable truncation of APC, AAPC-associate mutations do not appear to lead to the predicted truncated protein.
Fodde, R and Khan, P. Genotype-phenotype correlations at the adenomatous polyposis coli (APC) gene. Crit Rev Oncog 6 (1995) 291-303. Abstract
II. Hereditary Nonpolyposis Colorectal Cancer
Hereditary Nonpolyposis Colorectal Cancer (HNPCC), also known as Lynch syndrome, is an autosomal dominant predisposition to the development of tumors in the proximal colon but also in the endometrium, small bowel, ureter, and renal pelvis.
HNCPCC is caused by mutations in mismatch repair genes, in MSH2, MLH1, and to a lesser extent in MSH6, PMS1, and PMS2.
Microsatellite instability MSI
This disease is characterized by a high rate of changes in random microsatellites in tumor DNA when compared with normal genomic DNA from the same individual.
The detection of MSI in tumor DNA is indicative of genetic instability resulting from loss of DNA mismatch repair function.
Mutator phenotype hypothesis
This observation has led many investigators to speculate on the way defects in mismatch repair might cause cancer. One hypothesis, termed the 'mutator phenotype' hypothesis, postulates that mismatch repair defects lead to mutation in other genes, including those known to play a role in the adenoma - carcinoma sequence.
Therefore, according to the 'mutator phenotype' hypothesis, the increased mutation rate is then the cause of accelerated tumorigenesis.
However, it is not clear whether the 'mutator phenotype' initiates tumorigenesis. Resistance to apoptosis could also provide a selective advantage to the MMR-deficient cell to allow clonal expansion. In this hypothetical scenario, the mutator phenotype plays a more relevant role in tumor progression rather than in initiation.
In HNPCC a broad spectrum of tissues is at risk for cancer. As the result of ascertainment bias and clinical selection of colorectal cancer, research has mainly concentrated on families affected primarily by this tumor type. However, this is not an essential requirement for the disorder. Recently, the clinical criteria have been revised by the international collaborative group on HNPCC. In the revised criteria diagnosis requires at least 3 relatives with an HNPCC-associated cancer, including colorectal, endometrial, small bowel, uretral, or renal pelvic cancer.
Mutation analysis
MSH2 and MLH1. In a minority of cases, germline PMS2 and MSH6 mutations have been found. The presence of multiple endometrial cancers in the family is a characteristic of MSH6 mutations. Notably, female carriers of MSH6 mutations have a relatively lower risk of colorectal cancer risk compared to carriers of mutations in MSH2 and MLH1, but have a much increased risk of endometrial cancer. Also, microsatellite instability (MSI) due to loss of MSH6 occurs mainly at mononucleotide repeats.
Wijnen J, de Leeuw W, Vasen H, van der Klift H, Møller P, et al, Cornelisse C, Morreau H, and Fodde R. (1999) Germline mutations in MSH6 are responsible for familial endometrial cancer as part of the HNPCC clinical manifestations. Nature Genet, 23:142-144 - Abstract.

The table shows that about 73% of female MSH6 mutation carriers developed endometrial tumors, while only about 30% of MSH2 or MLH1 mutation carriers did so. 17% of MSH6 mutation carriers developed colorectal cancer, which is very low compared to 52% or 56% in carriers of MSH2 or MLH1 mutations. This observation does not hold true for males, in whom there is an identical incidence of colorectal cancer for carriers of mutations in MSH6, MSH2 and MLH1.
Wijnen JT, Vasen HF, Khan PM, Zwinderman AH, van der Klift H, Mulder A, Tops C, Møller P, Fodde R. (1998) Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med 339:511-8. - Abstract
Deletions
During the last decade PCR has become such a dominant method in mutation analysis that Southern blotting and other methods to detect genomic deletions have been somewhat neglected. Large deletions are not generally detectable by PCR, but account for about 25% of the total HNPCC.
Wijnen J, van der Klift H, Vasen H, Khan PM, Menko F, Tops C, Meijers Heijboer H, Lindhout D, Moller P, Fodde R MSH2 genomic deletions are a frequent cause of HNPCC. Nat Genet 1998 Dec;20(4):326-8. - Abstract
Mutation analysis in familes not complying with clinical criteria.
We are still faced with the problem of determining the cause of disease in a large number of families showing clustering for colorectal cancer but which do not comply to the revised criteria. What is the best way to screen these families for mutations?
Two signal transduction pathways are known to play a rate-limiting role in colorectal cancer. These are the TGF-b signal transduction pathway and the WNT signal transduction pathway, complex pathways with multiple involved proteins. In fact, each of the members of these signal transduction pathways is a potential candidate for genetic predisposition to colorectal cancer.
We are presently developing what we call 'tumor chips' or tissue microarrays. Hundreds tumor sections from large cohorts of patients can be spot in one slide and analysed by immunohistochemistry.
By using antibodies which recognize specific members of the signal transduction pathway we can rapidly screen large numbers of patients for loss of expression in tumor tissue. Mutation analysis of the corresponding patient for that particular gene will hopefully lead to the identification of the disease-causing mutation.